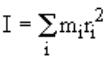
La determinazione della struttura
molecolare:
spettri
rotazionali e vibrazionali
I livelli energetici di una molecola “scaturiscono” dalla propria rotazione globale, dalle vibrazioni relative degli atomi e dai cambi nella sua configurazione elettronica.
La presenza delle righe spettrali di origine molecolare č spiegabile allo stesso modo delle righe spettrali atomiche: quando una molecola (o atomo) transisce da uno stato contraddistinto da un certo valore di energia a uno stato inferiore, il surplus di energia viene emesso sotto forma di un fotone avente una precisa frequenza. Esistono tuttavia delle sostanziali differenze fra spettroscopia molecolare e spettroscopia atomica dovute alla maggiore varietŕ dei modi in cui puň variare l’energia delle molecole rispetto agli atomi isolati. Infatti l’energia globale di un sistema molecolare non muta solo in funzione delle transizioni elettroniche da un orbitale all’altro, bensě anche a seguito di modifiche negli stati vibrazionali e rotazionali della molecola.
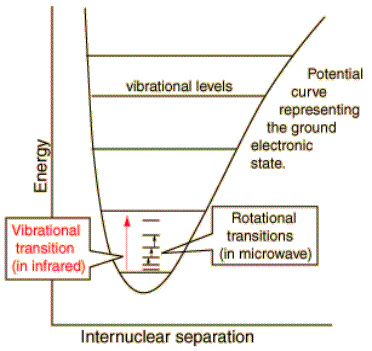
Analizzando uno spettro molecolare si possono ottenere informazioni riguardanti l’energia e le distanze di legame della molecola e quindi anche la sua forma. Tuttavia, mentre č possibile ottenere uno spettro rotazionale puro, lo spettro vibrazionale comprende anche righe dovute alle contemporanee variazioni di natura rotazionale.
Sperimentalmente esistono tre diversi modi per ottenere spettri molecolari:
1. La Spettroscopia di emissione viene osservata quando una molecola transisce da un certo stato energetico E” ad uno inferiore E’ (energia inferiore) emettendo l’energia corrispondente alla differenza fra i livelli sotto forma di un fotone avente una frequenza n coincidente con
n = (E” - E’)/h
dove h = 6.626 · 10-34 J·s indica la costante di Plank.
2. La Spettroscopia
di assorbimento consiste nell’osservare le frequenze in corrispondenza delle
quali la radiazione incidente č assorbita dal materiale in esame. Se per esempio la
molecola si trova nel piů basso livello energetico possibile (stato fondamentale), e
assorbe una radiazione di lunghezza d’onda l
= 470 nm (l = c/n,
con c velocitŕ della luce), allora esiste uno stato eccitato accessibile
corrispondente a 2.6 eV ( 21000 cm-1 se in funzione del numero d’onda s = 1/l) al di sopra di quello fondamentale.
3. La Spettroscopia
Raman studia i livelli energetici molecolari esaminando la luce diffusa dalle
molecole. Quando la collisione fra un raggio di luce incidente, di determinata frequenza, e le molecole del materiale provoca la diffusione,
compaiono nuove frequenze causate dall’acquisto o perdita di energia da parte del
fotone durante la collisone. Infatti se la luce incidente eccita le molecole, i fotoni
perderanno parte della loro energia e la luce diffusa emergerŕ con frequenze minori dell’originale.
Se al contrario, prima che vengano colpite dalla luce le molecole sono giŕ eccitate, esse
possono cedere energia ai fotoni incidenti che quindi compariranno nella radiazione
diffusa sotto forma di luce a frequenza superiore.
Livelli energetici e spettri
rotazionali
In un campione in fase gassosa, le molecole sono in continuo movimento dovutoto all’energia termica. Parte di questa si manifesta sotto forma di energia cinetica rotazionale associata appunto alle rotazioni relative ad un osservatore solidale al sistema di riferimento di laboratorio.
Per una singola e isolata molecola, questo moto č quantizzato in accordo con la teoria quantistica e le transizioni fra tali livelli rotazionali danno luogo allo spettro rotazionale della molecola in esame. Gli stati quantici di una molecola rimangono ben definiti quando essa č isolata, infatti a causa di collisioni con altre molecole essa puň cambiare il suo stato rotazionale perturbando e modificando il suo spettro.
A patto che il tempo fra le collisioni sia lungo rispetto all’inverso della frequenza di transizione (dell’ordine di 10-10 s), lo spettro rotazionale sarŕ nettamente definito. Questa condizione č facilmente realizzata nei gas a bassa pressione, meno di 10-4 atmosfere, infatti nei gas a piů alta pressione come nei liquidi, la frequenza delle collisioni č cosě elevata che le righe spettrali risultano tanto larghe da non poter distinguere le singole transizioni. Nello stato solido, l’orientamento delle singole molecole č pressoché fisso nello spazio e l’energia non puň essere “immagazzinata” sotto forma di energia rotazionale. Di conseguenza, la spettroscopia rotazionale č usata per studiare gas a bassa pressione cercando di limitare al minimo l’allargamento di riga dovuto ad una pressione troppo elevata e all’effetto Doppler.
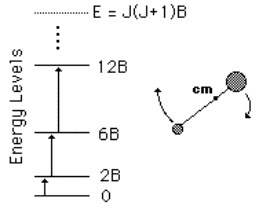
I livelli energetici rotazionali si possono ottenere risolvendo l’equazione di Scrodinger per la molecola oppure seguendo un approccio classico e quantizzandone il risultato.
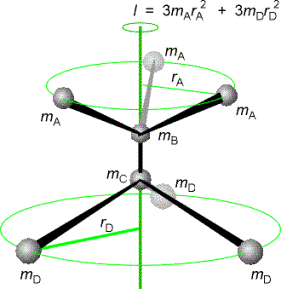
Il parametro chiave per descrivere gli spettri rotazionali č il Momento d’Inerzia della molecola.
Esso č definito come la sommatoria della massa di ogni atomo moltiplicata per il quadrato della sua distanza dall’asse di rotazione passante per il centro di massa della molecola:
dove ri č la distanza (ortogonale) dell’i-esimo atomo dall’asse di rotazione (Vedi figura a lato).
Poiché il momento d’inerzia dipende da tutte le masse e dalla geometria della molecola si intuisce facilmente come la spettroscopia rotazionale da informazioni riguardo gli angoli e le distanze di legame. In generale, le proprietŕ rotazionali di ogni molecola possono essere espresse in termini del momento d’inerzia attorno a tre assi perpendicolari scelti nella molecola. La convenzione č di chiamare i momenti d’inerzia Ia, Ib, e Ic, con gli assi scelti in modo che Ic ł Ib ł Ia (vedi figura in basso).
Per le molecole lineari il momento d’inerzia
attorno all’asse internucleare (congiungente i nuclei) č zero, poiché r č zero. La tabella allegata indica il 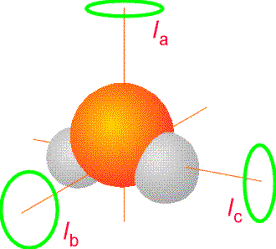 momento d’inerzia per molti
differenti tipi di molecole.
momento d’inerzia per molti
differenti tipi di molecole.
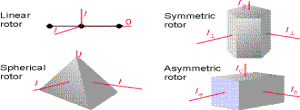
Considereremo per semplicitŕ che tutte le molecole
siano rotatori rigidi cioč che non cambino le mutue
distanze fra due punti fissati durante e non la rota-
zione. I rotatori rigidi possono essere classificati in
quattro differenti classi:
Rotore sferico; rotore simmetrico;
rotore lineare;
rotore asimmetrico.
Classicamente, l’energia di un corpo rigido rotante intorno ad un certo asse a si esprime mediante la relazione:
![]()
dove Ia č appunto il momento d’inerzia del corpo rigido e wa č la velocitŕ angolare intorno all’asse scelto misurata in radianti al secondo. Un corpo rigido libero di ruotare intorno ai tre assi ha ovviamente un’energia rotazionale totale data dalla:
![]()
Il momento angolare classico di un corpo avente momento d’inerzia Ia e velocitŕ angolare wa č:
![]()
di conseguenza, l’energia in funzione del momento angolare intorno a ciascun asse risulta:
![]()
Da questa equazione ricaveremo i livelli energetici rotazionali per i rotatori rigidi esclusi quelli asimmetrici il cui calcolo č noioso e complicato.
Livelli energetici di
un rotatore sferico
Per un rotatore sferico, come ad esempio la molecola del metano (CH4), il valore del momento d’inerzia intorno ai tre assi ortogonali coincide e l’espressione classica dell’energia si riduce a:
E = (1/ 2I) (Ja2
+ Jb2 + Jc2) = J2/2I
con J2 quadrato della grandezza (vettore) classica momento angolare totale. La corrispondente espressione quantistica si ottiene considerando che il momento angolare totale si limita ad assumere i valori
![]()
dove h coincide con h/2p. Ne risulta che l'energia di una molecola tipo rotatore sferico puň assumere i valori:
![]()
Il fattore h2 / 2I viene detto costante rotazionale della molecola e si indica con B, per cui:
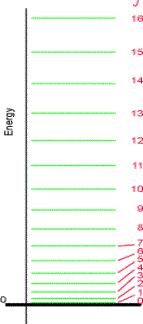
EJ = BJ(J + 1)
J = 0,1,2,….
L’intervallo energetico fra due livelli rotazionali adiacenti risulta:
EJ – EJ -1 =
2BJ
 E’ importante notare che la costante
rotazionale B decresce all’aumentare del momento d’inerzia della
molecola, di conseguenza i livelli energetici rotazionali saranno piů vicini per
le molecole di grandi dimensioni. Come esempio consideriamo appunto il caso del metano:
dalla lunghezza dei legami e dalla massa degli atomi si ottiene un momento d’inerzia
di 4.85 x 10-45 Kg m2, per cui B = 1.15 x 10-24 joule,
ovvero 0.058 cm-1. L’intervallo energetico č dell’ordine di 1.2 cm-1
quando J č circa 10, il che corrisponde a fotoni della regione spettrale delle microonde.
Anche per gli altri tipi di molecole l’ordine di grandezza dell’energia č piů
o meno uguale, ed č per questo motivo che lo studio delle transizioni rotazionali pure
costituisce un ramo della spettroscopia a microonde.
E’ importante notare che la costante
rotazionale B decresce all’aumentare del momento d’inerzia della
molecola, di conseguenza i livelli energetici rotazionali saranno piů vicini per
le molecole di grandi dimensioni. Come esempio consideriamo appunto il caso del metano:
dalla lunghezza dei legami e dalla massa degli atomi si ottiene un momento d’inerzia
di 4.85 x 10-45 Kg m2, per cui B = 1.15 x 10-24 joule,
ovvero 0.058 cm-1. L’intervallo energetico č dell’ordine di 1.2 cm-1
quando J č circa 10, il che corrisponde a fotoni della regione spettrale delle microonde.
Anche per gli altri tipi di molecole l’ordine di grandezza dell’energia č piů
o meno uguale, ed č per questo motivo che lo studio delle transizioni rotazionali pure
costituisce un ramo della spettroscopia a microonde.
Livelli energetici di
un rotore simmetrico
Nei rotori simmetrici, molecole tipo
ammoniaca (NH3), due momenti d’inerzia sono uguali ma differenti dal terzo
Ia = Ib ą Ic,
come per un cilindro. L’unico asse di simmetria della molecola č il suo asse
principale, il momento d’inerzia attorno a tale asse lo denoteremo con ![]() e
i due momenti d’inerzia eguali con
e
i due momenti d’inerzia eguali con ![]() . Se
. Se ![]() >
>
![]() il rotore č classificato come “oblate”
(schiacciato ai poli come il benzene C6H6), viceversa verrŕ
chiamato “prolate” (allungato come il cloruro di metile CH3Cl). Con
queste considerazioni l’espressione classica dell’energia per tale molecola
diventa:
il rotore č classificato come “oblate”
(schiacciato ai poli come il benzene C6H6), viceversa verrŕ
chiamato “prolate” (allungato come il cloruro di metile CH3Cl). Con
queste considerazioni l’espressione classica dell’energia per tale molecola
diventa:
![]()
espressione che puň essere riscritta in termini del quadrato del modulo del momento angolare
![]()
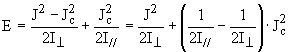
L’espressione quantica si ottiene effettuando la “consueta” sostituzione discreta:
![]()
Sappiamo inoltre dalla meccanica quantistica che le componenti del momento angolare lungo un qualunque asse sono quantizzate e limitate a valori interi multipli di h; di conseguenza il momento angolare intorno l’asse principale (asse c) č circoscritto ad assumere valori Kh con K = 0, ± 1, ± 2, ..., ± J. Nell’espressione dell’energia, quindi, sostituiamo a Jc la quantitŕ Kh, ottenendo :
E(J,K) = BJ(J+1) +(A – B) K2
Con
A = h2/
2![]() e
B = h2/
2
e
B = h2/
2![]() .
.
Notiamo che adesso l’energia
rotazionale dipende da due numeri quantici J e K 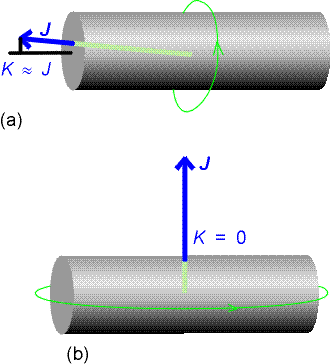 poiché ci sono
due distinti momenti d’inerzia per la molecola. L’importanza di K č dovuta al
fatto che l’energia di rotazione dipende dalla particolare distribuzione del momento
angolare totale. Quando K = 0 non c’č componente del momento angolare lungo l’asse
principale (vedi figura (b)), il moto assume interamente la caratteristica del testacoda
e l’energia dipende solamente da
poiché ci sono
due distinti momenti d’inerzia per la molecola. L’importanza di K č dovuta al
fatto che l’energia di rotazione dipende dalla particolare distribuzione del momento
angolare totale. Quando K = 0 non c’č componente del momento angolare lungo l’asse
principale (vedi figura (b)), il moto assume interamente la caratteristica del testacoda
e l’energia dipende solamente da ![]() .Se invece K = ± J la
rotazione molecolare ha luogo maggiormente intorno all’asse di simmetria (vedi figura
(a)) e i livelli energetici dipendono quasi interamente da
.Se invece K = ± J la
rotazione molecolare ha luogo maggiormente intorno all’asse di simmetria (vedi figura
(a)) e i livelli energetici dipendono quasi interamente da ![]() .
.
Si noti inoltre che il segno di K non influisce sull’energia (dipende infatti da K2) perché opposti valori di K corrispondono a sensi opposti nella rotazione della molecola e l’energia č indipendente dalla direzione della rotazione.
Livelli energetici di
un rotore lineare (molecole biatomiche)
Per i rotori lineari, come le molecole biatomiche dell’anidride carbonica (CO2), la rotazione avviene solamente intorno ad un asse ortogonale alla congiungente i nuclei e il momento angolare riferito a tale congiungente č nullo poiché č nullo il momento d’inerzia sempre riferito a tale asse. Di conseguenza la componente del momento angolare intorno all’asse principale di un rotore lineare č identicamente nulla e sarŕ nullo anche K. I livelli energetici di una molecola biatomica saranno allora dati dalla seguente espressione:
E(J) = BJ(J+1)
J = 0,1,2….
Dove la costante rotazionale della
molecola B = h2 / 2I con I unico momento d’inerzia
della molecola (![]() ).
).
Tale espressione dell’energia č identica a quella del rotore simmetrico ma qui K = 0 mentre per il rotore sferico l’espressione finale č dovuta al fatto che A = B.
Regole di selezione per le transizioni
rotazionali
La principale regola di selezione per l’osservazione
di un puro spettro rotazionale č che la molecola abbia un momento di dipolo
permanente. Poiché all’origine di un’onda elettromagnetica di frequenza n si puň sempre pensare che ci sia un dipolo elettrico
oscillante alla medesima frequenza, solamente le molecole dotate di momento dipolare
elettrico permanente sono suscettibili di emettere o di assorbire la radiazione subendo
una transizione fra stati rotazionali differenti. Le molecole dotate di un momento di
dipolo elettrico permanente sono chiamate polari ed interagiscono (si
accoppiano), al contrario di quelle non-polari, con un campo elettromagnetico
attraverso il 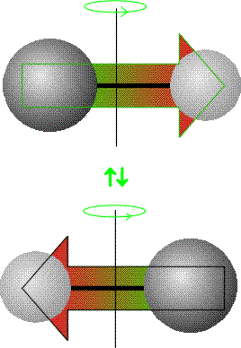 campo generato dalla propria rotazione (del tutto analoga ad un
dipolo oscillante).
campo generato dalla propria rotazione (del tutto analoga ad un
dipolo oscillante).
Questo concetto č mostrato nella figura a lato dove per un osservatore, una molecola polare rotante sembra un dipolo oscillante capace di interagire con un campo elettromagnetico.
Non si avrŕ dunque uno spettro rotazionale puro per tutte le molecole biatomiche omonucleari (O2, N2) e per i rotori lineari che sono definiti pertanto rotazionalmente inattivi. I rotori sferici invece sono rotazionalmente inattivi se non vengono “distorti” da forze rotazionali che vanno a modificare la loro distribuzione di carica.
Per calcolare le variazioni ammissibili
nel valore di J e K pensiamo al fatto che il fotone č dotato di momento angolare
di spin, e quando viene espulso o assorbito dalla molecola, nel corso di una transizione
rotazionale, il momento angolare della molecola deve mutare adeguatamente per compensare
quello fotonico (conservazione del momento angolare globale di un sistema). Essendo
unitario il momento angolare di spin del fotone, anche la conseguente variazione del
momento angolare 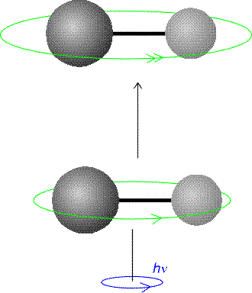 molecolare che accompagna la transizione dovrŕ essere unitaria.
La regola di selezione sarŕ quindi:
molecolare che accompagna la transizione dovrŕ essere unitaria.
La regola di selezione sarŕ quindi:
DJ = ± 1
La transizione DJ = + 1 corrisponde all’assorbimento di un fotone da parte della molecola (figura a lato) se sta ruotando nello stesso senso dello spin del fotone incidente. Invece la transizione DJ = - 1 corrisponde all'emissione di un fotone. Per i rotori simmetrici, poiché la luce non č in grado di accelerare la rotazione intorno al loro asse di simmetria, esiste un’ulteriore regola di selezione:
DK = 0
E’ da sottolineare inoltre che l’intensitŕ della radiazione emessa o assorbita č direttamente proporzionale al quadrato del momento dipolare elettrico permanente quindi, le molecole polari con momento piů elevato daranno uno spettro con righe piů intense di quelle delle molecole polari con momento minore.
Applicando le regole di selezione descritte all’espressione dell’energia, si possono calcolare le frequenze di assorbimento permesse per le transizioni J + 1 ? J :
E = hn = 2B (J + 1) J = 0,1,2,....
Lo spettro risulta essere suddiviso da una serie di righe di frequenze successive 2B, 4B, 6B….tutte separate di 2B (vedi figura seguente). Misurando tale distanza fra le righe (calcolato cioč B), si ricava il momento d’inerzia perpendicolare all’asse di simmetria della molecola e, note le masse degli atomi presenti nella molecola, č facile risalire alle informazione riguardanti quest’ultima come distanze, angoli di legame e quindi alla sua forma, anche se manca il valore dell’altro momento d’inerzia il cui calcolo puň essere sviluppato con altri metodi.
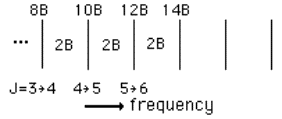
Intensitŕ e larghezza delle righe
spettrali
Analizzando gli spettri di diverse molecole ci si rende conto che l’intensitŕ delle righe č la piů varia. Una certa molecola possiede livelli energetici separati da uno specifico DE, ma capita chela frequenza associata a tale transizione non si rivela; questo perché l’intensitŕ delle righe dipende dal numero delle molecole che si trovano nei diversi stati al momento in cui si registra lo spettro e di quale sia la “predisposizione” delle singole molecole di assorbire o emettere fotoni. Sappiamo dalle regole di selezione che DJ = ± 1, ma č piů probabile la transizione J = 0 ® J = 1 o la transizione J = 4 ® J = 5 ? La meccanica quantistica “insegna” che tutte le transizioni con un uguale variazione di J sono equiprobabili tuttavia, come giŕ detto, questo non significa che tutte le righe di uno spettro rotazionale sono ugualmente intense. Esistono due fattori che determinano e influenzano l’intensitŕ di tali righe: la popolazione e la degenerazione caratteristiche di ogni livello energetico.
La popolazione indica il numero di molecole che si trovano in quel determinato stato energetico; per un campione di gas alla temperatura assoluta T ed avente un numero di molecole N, la porzione di molecole aventi energia EJ, č data dalla statistica di Boltzmann. E’ noto che l’energia rotazionale del piů basso stato di una molecola č zero poiché J = 0, quindi se abbiamo N0 molecole nel piů basso stato, il numero di molecole NJ in uno stato piů alto J, č dato da:
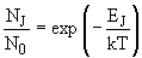
Per capire meglio, consideriamo un campione a temperatura ambiente (T = 300 K), che corrisponde ad un energia termica kT = 200 cm-1; i livelli molecolari rotazionali distano fra di loro solitamente di una frazione di cm-1 e anche a temperatura ambiente sono in parecchi ad essere popolati. La conseguenza ovvia č che le righe componenti lo spettro di assorbimento rotazionale, si devono a transizioni delle molecole originanti da una serie di stati rotazionali popolati. Possiamo graficare la relativa popolazione in funzione di J per una data temperatura e per valori diversi della costante rotazionale B:
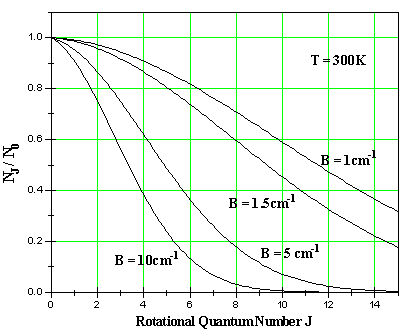
La degenerazione (dei livelli)
consiste nell’avere diversi livelli energetici caratterizzati dalla stessa energia,
di conseguenza una transizione fra tali livelli non implica un’emissione o
assorbimento di radiazione. Dalla meccanica quantistica č noto che per un dato valore del
momento angolare totale J, ci sono (2J + 1) possibili valori per MJ. Ogni
livello energetico rotazionale ha (2J + 1) livelli della stessa energia. Cioč, lo stato
rotazionale J = 0 č un livello energetico singolo (singoletto), lo stato J = 1 č
un triplo livello energetico (tripletto) e cosě via. Quindi sebbene la popolazione
molecolare di ogni livello energetico rotazionale diminuisce esponenzialmente con l’aumentare
di J, il numero dei possibili livelli aumenta 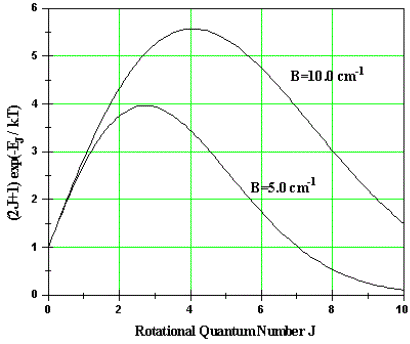 linearmente con J. La popolazione totale relativa di
un livello con energia EJ diventa quindi:
linearmente con J. La popolazione totale relativa di
un livello con energia EJ diventa quindi:
![]()
La figura a lato mostra appunto la popolazione relativa, tenendo conto della degenerazione dei livelli, in funzione di J per due differenti valori di B. Si noti che la curva presenta un massimo per un certo valore di J; tale massimo č naturalmente funzione della temperatura.
In conclusione possiamo affermare che l’intensitŕ delle righe spettrali č proporzionale alla temperatura di lavoro, alle popolazioni dei livelli energetici rotazionali e alla degenerazione di tali livelli. Transizioni fra livelli con valori molto bassi o molto alti di J, saranno caratterizzate da una riga spettrale meno intensa rispetto a quelle per cui il valore di J presenta un massimo (come in figura).
Le varie righe dello spettro hanno inoltre una differente larghezza.
Uno dei fenomeni responsabile di ciň č l’effetto Doppler ; quando una sorgente di radiazioni di lunghezza d’onda l si allontana dall’osservatore con velocitŕ v, l’osservatore rivela una radiazione con lunghezza d’onda (1 + v/c) l, dove c rappresenta la velocitŕ della luce. Una parte delle molecole del campione in esame si allontanano dall’osservatore, altre si avvicinano, alcune si muovono velocemente, altre lentamente. Come conseguenza si ha che la forma della riga rivelata rappresenta il profilo di assorbimento o di emissione che scaturisce dall’insieme degli spostamenti per effetto Doppler verificatisi nel campione. Poiché la percentuale di molecole che presentano una determinata velocitŕ č data dalla distribuzione di Maxwell, la cui curva č una gaussiana, la forma di una riga Doppler č data appunto da una curva gaussiana i cui parametri (larghezza e altezza) variano molto con la temperatura perché varia con essa la distribuzione delle velocitŕ molecolari (vedi figura). La larghezza a metŕ altezza di una riga risulta essere Dl = 2(l c) (2kT ln 2/m)1/2.
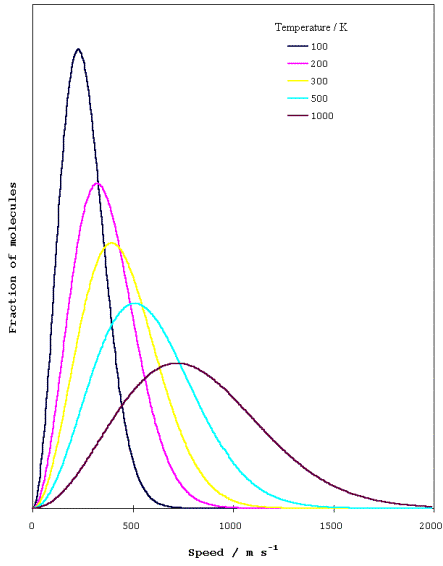
Per ottenere delle righe di nitidezza massima č opportuno operare a bassa temperatura ed eliminare cosě l’allargamento Doppler. Ma anche quando questo č divenuto trascurabile, agisce un altro fenomeno chiamato allargamento naturale. Esso dipende dal tempo di vita del livello energetico. Se lo stato del sistema varia alla velocitŕ 1/t, i suoi livelli energetici appaiono indistinti in una misura dell’ordine di dE dove dE ~ h /t. Cioč se una molecola rimane nel suo stato eccitato per un tempo uguale a t, la sua energia risulta E ± dE. Solo nel caso in cui lo stato in esame avesse vita infinitamente lunga sarebbe possibile determinarne con esattezza l’energia corrispondente. Da ciň emerge che le righe spettrali si estendono sopra un intero arco di frequenze e gli stati eccitati a vita breve sono caratterizzati da righe larghe, quelli a vita lunga da righe piů strette.
Livelli energetici e spettri
vibrazionali
Consideriamo per semplicitŕ una molecola biatomica, le cui vibrazioni si riducono unicamente allo stiramento (stretching) e alla contrazione dei legami. Nella formazione di una molecola stabile, si verifica un bilanciamento di forze, repulsiva fra la carica positiva dei nuclei atomici e fra ogni coppia di elettroni, attrattiva fra il nucleo di un atomo e gli elettroni dell’altro. I due atomi si stabilizzano ad una media distanza internucleare alla quale si ha un bilanciamento delle forze e l’energia totale del sistema presenta un minimo: il sistema č in equilibrio. Spingendo con forza gli atomi per stare piů vicini, la forza repulsiva subirŕ un rapido aumento, al contrario cercando di allontanare gli atomi si avrŕ una resistenza da parte della forza attrattiva. In entrambi i casi per distorcere il legame c’č bisogno di energia. E’ possibile graficare l’energia in funzione della distanza internucleare come nel seguente grafico in cui, considerata la molecola biatomica dell’acido cloridrico ( o cloruro di idrogeno, HCl), l’atomo di cloro č fissato all’asse delle ordinate ed immaginiamo di spingere e tirare l’atomo d’idrogeno piů vicino o piů lontano (dalla distanza di equilibrio). Maggiore č la forza impressa in un verso o nell’altro, maggiore risulta l’incremento energetico il cui valore risulta nullo solo nella posizione di equilibrio corrispondente alla distanza di legame della molecola:
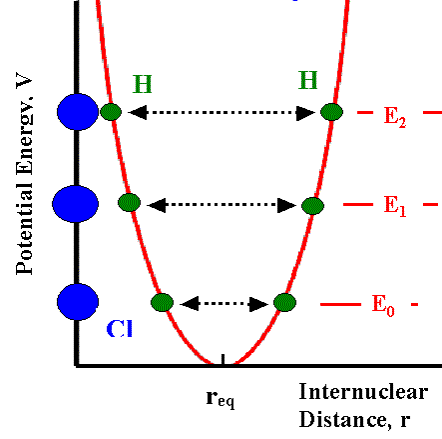
Tale grafico mostra appunto una tipica curva dell’energia potenziale di una molecola biatomica. Nelle regioni vicine a req (minimo della curva), l’energia potenziale puň essere approssimata ad una parabola, quindi possiamo scrivere:
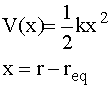
dove k rappresenta la costante elastica di forza del legame. Piů ripido č il potenziale (piů rigido č il legame), piů elevato č il valore della costante elastica. Essendo questo potenziale coincidente con quello di un oscillatore armonico semplice, nella molecola deve persistere un’oscillazione armonica semplice intorno alla posizione di equilibrio del legame ed inoltre i livelli energetici vibrazionali permessi risultano:
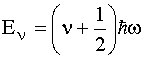 con
con 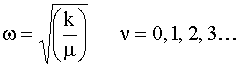
dove m č la massa ridotta (o massa effettiva) della molecola uguale a:
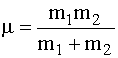
Perň tali livelli energetici non sono
che un’approssimazione dei veri livelli, in quanto dovuti all’approssimazione
parabolica della curva dell’energia potenziale; infatti quando l’energia delle
vibrazioni č tale da imporre agli atomi un considerevole spostamento dalla posizione di
equilibrio del legame, l’approssimazione parabolica perde di significato: essa non
consente di dissociare la molecola nei suoi atomi componenti ne di avvicinarli
indefinitamente. Alle alte frequenze vibrazionali i contorni del potenziale risultano meno
ristretti e ci si attende che ad alta energia, gli intervalli fra i livelli energetici
vibrazionali siano minori che a bassa energia (vedi figura seguente); in tale situazione
le oscillazioni degli atomi consentono alla molecola di “esplorare” regioni
della curva dell’energia potenziale dove l’approssimazione parabolica come giŕ
detto perde di significato e sono richiesti 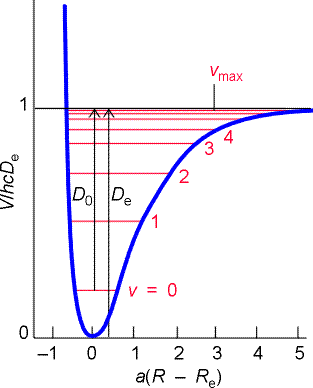 termini addizionali per descrivere tale stato.
termini addizionali per descrivere tale stato.
Il moto vibrazionale diventa quindi anarmonico e una forma matematica che si accosta bene alla reale curva dell’energia potenziale č stata suggerita da Morse, il cui potenziale risulta:
![]()
dove a č
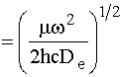
e De rappresenta la profonditŕ del minimo dell’energia potenziale. D0 rappresenta l’energia di dissociazione della molecola cioč, l’energia necessaria per rompere il legame molecolare partendo da una molecola biatomica nello stato vibrazionale n = 0. L’espressione energetica dei livelli dovuti ad un tale potenziale diventa:
En = (n + ˝)
hw
– (n + ˝)2
xe hw
In cui la costante di anarmonicitŕ xe č uguale a:
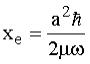
Il fatto che il secondo termine venga sottratto dal primo conferma il previsto convergere dei livelli energetici vibrazionali all’aumentare dell’energia (numeri quantici vibrazionali).
Regole di selezione per le transizioni
vibrazionali
Lo stiramento o il piegamento dei legami molecolari cui origina la vibrazione della molecola possono o meno far variare il momento di dipolo posseduto dalla stessa molecola. Tale dipolo oscillante (piů o meno armonicamente) consente l’emissione o l’assorbimento di radiazione elettromagnetica. Ci sono tuttavia delle vibrazioni che non alterano il momento di dipolo molecolare, come il moto di stretching nelle molecole biatomiche omonucleari, non interagendo di conseguenza con la radiazione e neanche generandola.
Infatti lo stiramento del legame fra gli atomi di una molecola biatomica omonucleare non altera il valore (nullo) del momento dipolare, quindi le molecole biatomiche omonucleari non presentano alcuno spettro vibrazionale (infrared inactive). Al contrario nelle molecole biatomiche eteronucleari, il momento dipolare varia al variare della distanza di legame; di conseguenza tali molecole sono in grado di fornire uno spettro vibrazionale sia in assorbimento che in emissione (infrared active).
Quindi la piů evidente regola di selezione per le vibrazioni molecolari č che il momento di dipolo elettrico di una molecola deve cambiare quando gli atomi vengono spostati relativamente l’uno rispetto all’altro.
Tuttavia non č necessario che le molecole abbiano un momento di dipolo permanente diverso da zero per transire fra livelli vibrazionali permessi, infatti il loro momento dipolare puň passare da zero a un valore diverso da zero.
La regola di selezione specifica per un oscillatore armonico č :
D n
= ±
1
ottenuta dalla conservazione del momento angolare
applicata al sistema molecola+fotone. L’espressione dei livelli energetici risulta
quindi:
DE = hw
che tenendo conto dell’anarmonicitŕ diventa:
DE = hw - 2(n + 1) xe hw
- ...
L’energia in piů o in meno si manifesta sotto forma di un fotone la cui frequenza č tipica della regione infrarossa dello spettro della luce, da qui la denominazione di infrared active or inactive per le molecole che presentano o meno uno spettro vibrazionale. A temperatura ambiente, kT = 200 cm-1, la maggior parte delle molecole si trova nel proprio stato vibrazionale fondamentale, pertanto la transizione dominante, risulta in spettroscopia I.R. quella da n = 0 a n = 1.
Poiché le vibrazioni possono risultare lievemente anarmoniche, bisogna considerare che le energie di transizione dipendono in piccola misura da n e che viene infranta la regola di selezione (basata sul moto armonico) e diventano possibili transizioni con
D n
= ±
2
chiamate seconde
armoniche al pari delle transizioni con D n = ± 1 dette prime armoniche o fondamentali. Alla
seconda armonica corrispondono fotoni di energia:
DE = 2hw - 2(2n + 3) xe hw
- ... n + 2 ? n
La comparsa di righe che traggono origine da livelli differenti č utile al calcolo di xe e di De .
Spettri vibrorotazionali
Ogni linea di uno spettro vibrazionale di una molecola biatomica eteronucleare, effettuato ad alta risoluzione, consiste di un gran numero di righe strettamente intervallate. E’ per questo motivo che gli spettri molecolari sono chiamati spettri di bande (vedi figura in basso) mentre quelli atomici sono chiamati spettri di righe.
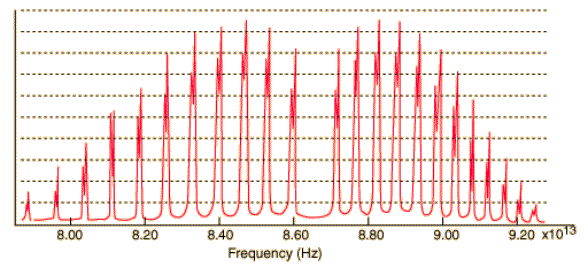 Gli intervalli
fra le righe di una stessa banda sono dell’ordine di qualche cm-1, e
questo significa che tale struttura sia dovuta a transizioni rotazionali contemporanee
alla transizione vibrazionale. Infatti la quantitŕ di energia che entra in gioco nella
transizione vibrazionale č talmente grande da eccitare i livelli rotazionali molecolari.
Gli intervalli
fra le righe di una stessa banda sono dell’ordine di qualche cm-1, e
questo significa che tale struttura sia dovuta a transizioni rotazionali contemporanee
alla transizione vibrazionale. Infatti la quantitŕ di energia che entra in gioco nella
transizione vibrazionale č talmente grande da eccitare i livelli rotazionali molecolari.
Inoltre la variazione vibrazionale č assimilabile a un mutamento transitorio del momento d’inerzia, al quale la molecola risponde con il mutamento del proprio stato rotazionale ed in particolare variando la frequenza del moto rotatorio per mantenere costante il valore del momento angolare totale della molecola. Per il principio di conservazione del momento angolare J = Iw, ad una diminuzione del momento d’inerzia deve corrispondere un aumento della frequenza angolare.
 L’indagine
particolareggiata dimostra che la regola di selezione per la variazione rotazionale
associata al mutamento vibrazionale č ancora:
L’indagine
particolareggiata dimostra che la regola di selezione per la variazione rotazionale
associata al mutamento vibrazionale č ancora:
DJ = ± 1
eccetto che in circostanze particolari, come nel caso della molecola di monossido di azoto (NO), in cui si registra un momento angolare elettronico intorno all’asse della molecola lineare e la conservazione del momento angolare a tale sistema, puň essere soddisfatta anche dalla condizione DJ = 0.
La figura a lato mostra la suddivisione dei livelli energetici molecolari, come si vede fra uno stato vibrazionale e l’altro esistono diversi stati rotazionale spaziati da distanze molto minori rispetto quelli vibrazionali. Di conseguenza l’espressione dei livelli energetici di una molecola soggetta a vibrazione e rotazione, tenuto anche conto del fattore di anarmonicitŕ, risulta:
E(J,n) = BJ(J+1) + (n + ˝) hw – (n + ˝)2 xe hw
Si noti che la costante rotazionale B č la stessa per tutti i valori sia di J che di n, la separazione fra due livelli di un dato J č la stessa sia nello stato n = 0 che in quello n = 1.
Un’analisi dettagliata della meccanica quantistica sui cambiamenti simultanei nelle rotazioni e vibrazioni di una molecola, mostra che il numero quantico rotazionale J cambia di ± 1 durante la transizione vibrazionale delle molecole biatomiche. La regola di selezione vibrazionale č:
D n
= ±
1 ± 2 ± 3
Quando si verifica una transizione vibrazionale non solamente n passa ad n + 1 (nell’assorbimento), ma J cambia di 0 (non cambia) o di ± 1, tale variazione dipende dalle molecole considerate.
La frequenza
n0 = n (1 - 2 xe)
č chiamata origine o centro di banda; essa individua la regione centrale della riga di assorbimento vibrazionale, la quale risulta suddivisa in tre insiemi di righe detti rami contrassegnati da tre diverse lettere in base al valore assunto da DJ.
Tale situazione č illustrata nella figura seguente dalla quale si evince inequivocabilmente la contemporanea eccitazione dei livelli rotazionali durante una transizione vibrazionale. Come si vede appunto dalla figura, per ogni livello vibrazionale esistono nove livelli rotazionali permessi.
Passiamo ora ad una piccola ma importante descrizione dei tre rami in cui č divisa la riga spettrale vibrazionale:
Ramo P: Tali righe corrispondono
a transizioni per le quali valgono D n = 1 e DJ = -1. Quindi se la 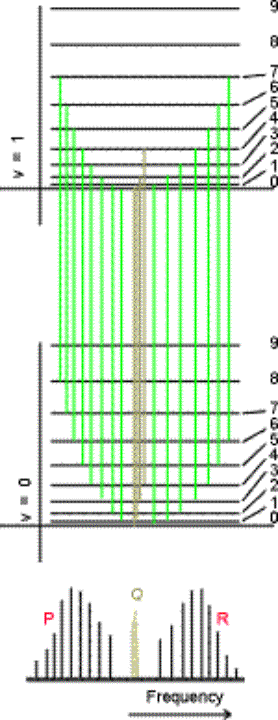 molecola si trovava inizialmente nello
stato J, la transizione č J ® J = -1 e il cambiamento totale di energia che
determina anche il valore delle frequenze di tali righe č (basta dividere per h):
molecola si trovava inizialmente nello
stato J, la transizione č J ® J = -1 e il cambiamento totale di energia che
determina anche il valore delle frequenze di tali righe č (basta dividere per h):
DE = hw - 2BJ
Ciň rende conto delle righe che cadono alla sinistra del centro di banda aventi delle frequenze inferiori coincidenti con: w – 2B, w – 4B,…
Ramo Q: Le righe di tale ramo prendono origine dalle transizioni con D n = 1 e DJ = 0. Le loro frequenze sono date dal valore energetico:
DE = hw
per tutti i valori di J. Il ramo Q, quando č permesso, č una sola riga alla frequenza di centro banda, ma nelle molecola reali, poiché le costanti rotazionali sono diverse per i diversi livelli vibrazionali, tale ramo risulta dall’addensarsi di un gruppo di righe strettissimamente intervallate (come si evince dalla figura qui a lato).
Ramo R: Risulta composto dalle righe le cui frequenze corrispondono alle transizioni con D n = 1 e DJ = 1 e si calcolano facilmente dalla seguente e relativa espressione dei livelli energetici:
DE = hw + 2B(J + 1)
Le righe sono spostate di 2B, 4B,… verso le alte frequenze rispetto alla fondamentale (frequenza di centro banda).
Dall’intervallo fra le righe P ed R di una transizione vibrazionale, si ricava il valore di B e da questo la lunghezza del legame senza bisogno di ricorrere allo spettro a microonde, rotazionale puro, anche se con quest’ultimo si ottengono risultati di una maggiore precisione.
Spettroscopia Raman
Nella spettroscopia Raman i livelli
energetici della molecola sono “esplorati” analizzando le frequenze presenti
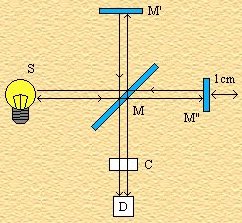
nella radiazione scatterata (diffusa) dalla molecola. In un tipico esperimento Raman, un
fascio di luce monocromatica viene fatto incidere sul campione per poi rilevarne la 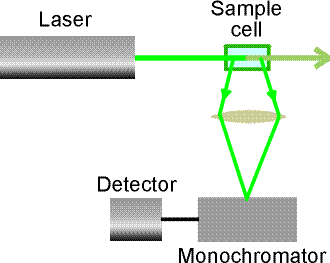 radiazione diffusa ad un angolo ortogonale rispetto alla direzione
del fascio incidente (vedi figura). Quando i fotoni incidenti urtano la molecola, essi
possono perdere o guadagnare energia: se la molecola assorbe parte dell’energia del
fotone per passare ad uno stato eccitato, il fotone riemergerŕ con un’energia
(frequenza) minore di quella d’incidenza andando a formare le cosiddette righe Stokes
dello spettro Raman; se invece la molecola si trova giŕ in uno stato eccitato (l‘energia
termica a temperatura ambiente eccita alcuni stati rotazionali) puň cedere energia al
fotone incidente che riemergerŕ dall’urto con un’energia maggiore andando a
formare le righe anti-Stokes.
radiazione diffusa ad un angolo ortogonale rispetto alla direzione
del fascio incidente (vedi figura). Quando i fotoni incidenti urtano la molecola, essi
possono perdere o guadagnare energia: se la molecola assorbe parte dell’energia del
fotone per passare ad uno stato eccitato, il fotone riemergerŕ con un’energia
(frequenza) minore di quella d’incidenza andando a formare le cosiddette righe Stokes
dello spettro Raman; se invece la molecola si trova giŕ in uno stato eccitato (l‘energia
termica a temperatura ambiente eccita alcuni stati rotazionali) puň cedere energia al
fotone incidente che riemergerŕ dall’urto con un’energia maggiore andando a
formare le righe anti-Stokes.
La componente della radiazione diffusa nella stessa direzione d’origine senza cambiamenti di frequenza č chiamata radiazione Rayleigh.
Gli spostamenti in frequenza della radiazione diffusa dalla radiazione incidente sono molto piccoli e di conseguenza la radiazione incidente deve essere “molto monocromatica” per poter osservare gli spostamenti in frequenza. Inoltre l’intensitŕ della radiazione diffusa č molto bassa quindi č necessario che il fascio incidente sia anche molto intenso. I Laser sono delle sorgenti di radiazioni elettromagnetiche perfetti per tali esperimenti. I primi spettri Raman compiuti con un laser vennero esaminati usando luce incidente con frequenze nel visibile e nell’ultravioletto, oggi la radiazione incidente cade nel vicino infrarosso per evitare complicazioni scaturenti dalla stimolazione della fluorescenza. La rivelazione della radiazione Raman č effettuata di solito mediante dei detector a semiconduttore; essa risulta complementare alla spettroscopia infrarosso perché , come vedremo fra poco, obbedisce a diverse regole di selezione e vengono osservate diverse transizioni.
Spettri Raman rotazionali
La piů importante regola di selezione per le transizioni Raman rotazionali č che la molecola abbia una polarizzabilitŕ anisotropa: una molecola quando č immersa in un campo elettrico viene distorta (subisce una polarizzazione per deformazione) a causa delle forze elettriche agenti sui nuclei in un senso e sugli elettroni in senso opposto. L’intensitŕ di tale deformazione dipende dalle caratteristiche fisico-chimiche della molecola rappresentate in tal senso da una grandezza che prende il nome appunto di polarizzabilitŕ elettrica (a). Una polarizzabilitŕ elevata porta la molecola a essere deformata anche da un campo elettrico esterno di lieve intensitŕ. Per esempio č piů facile deformare un atomo di Xenon, i cui elettroni esterni sono legati meno fortemente al nucleo, rispetto ad un atomo di Elio i cui unici due elettroni sono legati fortemente al nucleo; di conseguenza diremo che lo Xenon ha una polarizzabiltŕ piů elevata dell’Elio.
Piů precisamente, se l’intensitŕ del campo elettrico esterno applicato č E allora la molecola acquisisce un momento di dipolo indotto la cui ampiezza (o modulo) risulta:
![]()
che va ad aggiungersi a ogni momento di
dipolo permanente che abbia o meno la molecola. Un atomo č polarizzabile isotropicamente
poiché qualunque sia la direzione del campo elettrico applicato, č indotta la stessa
deformazione della nuvola elettronica. Per le molecole questo non č valido ad esclusione
ovvia dei rotori sferici, infatti le molecole non sferiche hanno una polarizzabilitŕ che
dipende dalla direzione del campo applicato alla molecola. Quindi tali molecole sono 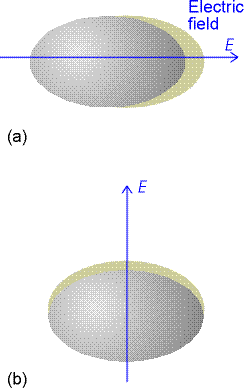 contraddistinte da avere una polarizzabilitŕ anisotropa. Per
esempio, come si vede nella figura, la distribuzione elettronica della molecola d’idrogeno
(H2) č piů deformata quando il campo elettrico č applicato parallelamente
alla congiungente i nuclei (direzione del legame) rispetto alla direzione ad essa
ortogonale, quindi avremo in tal caso:
contraddistinte da avere una polarizzabilitŕ anisotropa. Per
esempio, come si vede nella figura, la distribuzione elettronica della molecola d’idrogeno
(H2) č piů deformata quando il campo elettrico č applicato parallelamente
alla congiungente i nuclei (direzione del legame) rispetto alla direzione ad essa
ortogonale, quindi avremo in tal caso:
![]()
Tutte le molecole lineari e biatomiche, sia omo che eteronucleari, hanno una polarizzabilitŕ anisotropa e quindi sono rotazionalmente raman attive.Questa č una delle ragioni dell’importanza della spettroscopia raman, poiché tale tecnica puň essere usata per studiare la struttura rotazionale di molecole inaccessibile alla spettroscopia delle microonde (puri spettri rotazionali). Infatti la spettroscopia Raman puň essere usata per studiare la struttura rotazionale di molecole non possedenti un momento di dipolo permanente e quindi non mostrando uno spettro rotazionale nella regione delle microonde.
Regole di selezione per
le transizioni Raman
Negli spettri Raman rotazionali le regole di selezione specifica sono:
Rotori lineari: D J = 0, ± 2
Rotori simmetrici: D J = 0, ± 1,
± 2: D K
= 0
La presenza della cifra 2, quella
piů significativa, si puň ricondurre al fatto che ad ogni rivoluzione 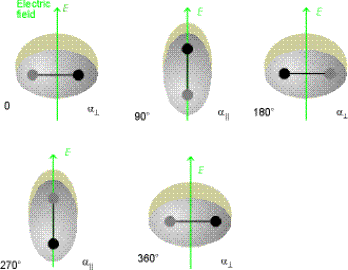 la
polarizzabilitŕ della molecola riacquista il valore iniziale due volte (vedi figura a
lato); considerando il momento di dipolo sembra che la molecola ruoti a velocitŕ doppia.
Inoltre considerando la conservazione del momento angolare e che lo spin fotonico ha
valore unitario, tale cifra resta giustificata dal fatto che nel processo Raman entrano in
gioco due fotoni, uno incidente e l’altro emergente.
la
polarizzabilitŕ della molecola riacquista il valore iniziale due volte (vedi figura a
lato); considerando il momento di dipolo sembra che la molecola ruoti a velocitŕ doppia.
Inoltre considerando la conservazione del momento angolare e che lo spin fotonico ha
valore unitario, tale cifra resta giustificata dal fatto che nel processo Raman entrano in
gioco due fotoni, uno incidente e l’altro emergente.
Classicamente il requisito della polarizzabilitŕ anisotropa e della presenza del fattore 2 č legata al fatto che se si irradia una molecola con una luce di frequenza wi, il momento di dipolo indotto nella molecola dipende dall’ampiezza del campo elettrico applicato:
![]()
Se la molecola ruota con frequenza angolare di rotazione wR, allora per un osservatore esterno la sua polarizzabilitŕ dipende anche dal tempo (se č appunto anisotropa) e coincide con:
![]()
dove a0 rappresenta la polarizzabilitŕ
media, ![]() e a varia da a
o + D a
a a o - D a mentre
la molecola ruota. Sostituendo l’espressione della polarizzabilitŕ nell’espressione
del momento di dipolo indotto ed eseguendo alcune relazioni trigonometriche, otteniamo:
e a varia da a
o + D a
a a o - D a mentre
la molecola ruota. Sostituendo l’espressione della polarizzabilitŕ nell’espressione
del momento di dipolo indotto ed eseguendo alcune relazioni trigonometriche, otteniamo:
![]()
![]()
![]()
Questi calcoli mostrano che il momento
di dipolo indotto ha una componente oscillante alla stessa frequenza della radiazione
incidente, questa componente come detto genera la componente Rayleigh. Il momento ha
inoltre due componenti oscillanti a wi
+ 2wR che danno la spiegazione
delle “spostate” righe Raman: il segno meno corrisponde alle righe Stokes,
il segno piů alle righe 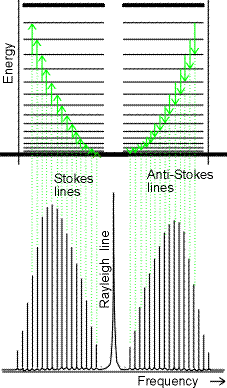 anti-Stokes (vedi figura a lato).
E’ importante notare come tali linee appaiono solamente se D a ? 0 e
cioč che la polarizzabilitŕ sia appunto anisotropa.
anti-Stokes (vedi figura a lato).
E’ importante notare come tali linee appaiono solamente se D a ? 0 e
cioč che la polarizzabilitŕ sia appunto anisotropa.
Quando la molecola subisce una transizione con D J = + 2 la luce diffusa lascia la molecola in uno stato rotazionale superiore (eccitato) e quindi ne emerge con frequenza (energia) diminuita. Tale transizione spiega la presenza delle righe Stokes dello spettro raman; la frequenza corrispondente puň essere ricavata, conoscendo i livelli energetici molecolari, dalla seguente espressione:
Righe Stokes (D J = + 2):
n = n0 – 2B(2J + 3)
per J
® J
+ 2
Tali righe compaiono sul lato di minore frequenza rispetto a quella della luce incidente n0, e con spostamenti 6B, 10B,14B,… In corrispondenza di stati ad elevato valore di J, l’intensitŕ delle righe si attenua poiché sono stati scarsamente popolati.
Quando invece la molecola, collidendo con il fotone incidente, subisce la transizione D J = - 2, il fotone acquisisce l’energia eccedente e riemerge con una frequenza superiore andando a formare le righe anti-Stokes, la cui determinazione in frequenza č data da:
Righe anti-Stokes (D J = - 2):
n = n0 + 2B(2J – 1)
per J
® J
– 2
Gli spostamenti in tale caso sono sul lato delle frequenze superiori rispetto a quella della luce incidente e valgono 6B, 10B, 14B,…
Dall’analisi dello spettro rotazionale Raman, ed in particolare dalla misura della spaziatura fra le righe, si ricavano informazioni riguardanti il momento d’inerzia (calcolato dal valore di B) della molecola perpendicolare all’asse di simmetria della stessa molecola e da questa misura i dati riguardanti il legame esistente nella molecola.
Spettri Raman vibrazionali
La regola di selezione generale per la comparsa dello spettro Raman vibrazionale si basa sulla variabilitŕ della polarizzazione elettrica molecolare. Solo se la vibrazione della molecola implica una variazione della polarizzabilitŕ, tale vibrazione molecolare č attiva nel Raman.
Il “solito” fotone incidente puň dar luogo a scambi d’energia se l’urto con la molecola provoca una eccitazione (diminuzione della frequenza del fotone incidente) o diseccitazione (aumento della frequenza del fotone) vibrazionale della molecola con conseguente variazione della polarizzabilitŕ. La regola di selezione specifica č:
D n = ± 1
Le righe che compaiono a frequenze superiori rispetto
la radiazione incidente, sono le anti-Stokes e corrispondono a D n = - 1. Si tratta di righe molto poco intense a
causa della scarsa popolazione di livelli vibrazionali eccitati. Le righe che si trovano
sul lato delle frequenze minori sono le Stokes, accompagnate da transizioni D n = + 1. Ad entrambi gruppi di righe si sovrappongono come
nel caso della spettroscopia infrarossa, la struttura ramificata derivante dalle
transizioni rotazionali in seno a quelle vibrazionali.
Per tali transizioni rotazionali vale la regola di selezione D J = 0 o D J = ± 2, come per gli spettri Raman rotazionali puri. I rami corrispondenti si dicono ramo O per D J = - 2, ramo Q per D J = 0 quando č permesso, e ramo S per D J = + 2.
Tale struttura č in totale analogia agli spettri vibrorotazionali puri (mostrati in figura a pagina 15); spettri ottenuti dall’assorbimento o dall’emissione di fotoni da parte della molecola, mentre nella spettroscopia Raman si analizza la radiazione diffusa dagli urti dei fotoni incidenti con la molecola in esame. In particolare la spettroscopia Raman offre la possibilitŕ di studiare le molecole biatomiche omonucleari (non dotate di momento di dipolo permanente) ricavando dalla spaziatura fra le righe Stokes (le anti-Stokes sono come detto troppo poco intense) la loro costante di forza misurante la rigiditŕ della molecola. Inoltre č possibile interpretare la struttura rotazionale degli spettri in funzione delle costanti rotazionali delle molecole per poter stabilire la lunghezza dei legami ecc.
Per le molecole poliatomiche esistono
dei modi vibrazionali attivi nel Raman ed altri no. La
regola di esclusione afferma che se la molecola possiede un centro di simmetria (il
centro di simmetria delle cariche positive coincide con quello delle cariche negative =
molecole apolari), allora i suoi modi che risultano inattivi nell’infrarosso sono
attivi nel Raman e viceversa.
Questa regola vale per l’anidride carbonica (CO2) ma non per l’acqua (H2O) mancando in tale molecola il centro di simmetria. In definitiva per avere tutti i dati relativi alla rigiditŕ (campo di forze) di una molecola, č necessario servirsi tanto dello spettro I.R. quanto del Raman.
Spettrometria
in trasformata di Fourier (FT-IR) |
Questa tecnica strumentale č basata sulla spettrometria
infrarossa classica. Si tratta di una tecnica recente creata grazie alla computerizzazione
del laboratorio strumentale. Contrariamente agli spettrometri tradizionali in questa
apparecchiatura non si ha un monocromatore a dispersione, ma viene utilizzato l’interferometro
di Michelson, il quale produce nel corso di una speciale scansione l’interferogramma
della sostanza in esame. Quest’ultimo č trasformato dal calcolatore collegato allo
strumento in un tradizionale spettro infrarosso mediante trasformata di Fourier. Inizialmente molto costosi e non molto precisi oggi
questi strumenti hanno in sostanza sostituito il tradizionale spettrofotometro a
dispersione. Una sorgente luminosa ad incandescenza emette un raggio
luminoso nel campo dell’IR, tale raggio giunge ad uno specchio semiriflettente
(beamsplitter) che lo divide in 50% ad uno specchio fisso ed un 50% allo specchio mobile.
I raggi riflessi da questi specchi sono inviati un’altra volta allo specchio
semiriflettente il quale ricongiunge i due raggi e li invia al rivelatore. Anche se i due
raggi hanno raggiunto il rivelatore congiunti essi hanno compiuto un diverso cammino
ottico: a seconda della differenza del cammino ottico dei due raggi si creano delle
interferenze costruttive o distruttive che creano un segnale al rilevatore proporzionale
alla differenza di cammino ottico dei due raggi e quindi dalla posizione dello specchio
mobile in quell’istante. In base al movimento del suddetto specchio tutte le
radiazioni monocromatiche contenute nella luce emessa dalla lampada danno luogo ad un
segnale complessivo di interferenza (interferogramma) che contiene in sé le
informazioni riguardanti la frequenza e l’intensitŕ della radiazione emessa. La
trasformata di Fourier, effettuata dal calcolatore dello strumento una volta che il raggio
č arrivato al detector, mostra sullo schermo un tradizionale spettro infrarosso,
trasformando il segnale di intensitŕ luminosa in funzione del tempo (spostamento dello
specchio) in segnale di intensitŕ in funzione del numero d’onda.
Un requisito fondamentale del FT-IR e quello di ottenere
un buon interferogramma, dal quale dipende la precisione dello spettro ottenuto, in altre
parole lo specchio mobile deve avere una velocitŕ costante e la sua posizione deve essere
nota in maniera esatta in ogni istante. Lo specchio deve inoltre mantenere una planaritŕ
costante durante tutto il suo spostamento Nella regione del lontano infrarosso (tra 200 e 10 cm-1)
gli spostamenti dello specchio avvengono tramite una vite micrometrica, mentre nella
regione del medio infrarosso gli spostamenti hanno bisogno di un meccanismo piů preciso e
raffinato: gli spostamenti dello specchio, sostenuti da cuscinetti d’aria in
manicotti d’acciaio a tenuta, avvengono tramite il passaggio di corrente (variabile)
in una bobina elettromagnetica, al termine della corsa lo specchio ritorna alla posizione
iniziale con un’inversione di corrente. Lo spostamento dello specchio varia da 1 a 20
cm con una velocitŕ di 0.01 e 10 cm/s. Per le analisi spettrofotometriche i campioni diluiti
sono molto piů utilizzati, poiché č possibile intervenire sia sulla concentrazione che
sul cammino ottico della cella in modo da operare nell’intervallo migliore d’assorbanza.
Purtroppo questa tecnica non puň essere molto utilizzata nell’IR poiché non
esistono solventi organici che siano trasparenti in tutta la regione dello spettro IR,
perciň vengono utilizzate, sia per i campioni liquidi che per quelli solidi, tecniche che
a volte possono complicare la determinazione accurata del campione. I solventi organici piů utilizzati per analisi IR sono
il tetracloruro di carbonio, il cloroformio, il solfuro di carbonio. Per i liquidi spesso
non esiste un solvente opportuno perciň si č costretti ad eseguire l’analisi sul
liquido puro. Per ottenere uno spettro soddisfacente, con un cammino ottico
sufficientemente corto, si usano quantitŕ minime di campione: di solito si pone una
goccia di campione fra due dischi di bromuro di potassio (KBr), i due dischi vengono poi
messi nel percorso del cammino ottico. Questa non dŕ risultati accettabili per un’analisi
quantitativa. I solidi spesso non sono solubili nei comuni solventi
usati per l’infrarosso, i campioni perciň si preparano per una dispersione del
campione in un liquido o in una matrice solida (mull). Per questa tecnica bisogna
ridurre il campione in polvere per non avere un’eccessiva assorbanza. L’alternativa piů comune consiste nel macinare
finemente il campione e mescolarlo ad un olio pesante (per es. olio di vaselina detto nujol),
altrimenti per non avere l’assorbanza dell’idrocarburo che potrebbero
interferire con l’assorbanza del campione viene usato un polimero fluorurato (fluorolube):
in entrambi i casi la mistura ottenuta viene pressata tra due finestre di alogenuro
alcalino. Il campione finemente polverizzato puň anche essere
mescolato a bromuro di potassio essiccato (che non dŕ assorbimento nell’infrarosso).
La miscela si ottiene mescolando il sale con l’analita in un mortaio, ed essiccando
poi la miscela sottovuoto ad alta pressione in modo da ottenere un disco trasparente da
porre nel cammino ottico del raggio. I campioni sia liquidi che solidi per essere alloggiati
nello strumento hanno bisogno di essere prima messi in un’apposita cella smontabile;
il campione viene posto fra due finestre di un alogenuro alcalino: fra i piů usati
cloruro di sodio o bromuro di potassio (NaCl, KBr). La finestra contenente il campione
trova alloggio nella cella smontabile: le piů sofisticate sono corredate anche di
appositi spessori per variare lo spazio di riempimento della finestra. Le finestre di alogenuro alcalino necessitano di essere
conservate in essiccatori perché non assorbano umiditŕ. La spettroscopia IR si basa sul principio dell’interazione
delle onde elettromagnetiche (in questo caso delle onde appartenenti al campo infrarosso)
con la materia. In particolare le radiazioni IR interferiscono con i legami interni alla
molecola ovvero con i legami fra i vari atomi e dei gruppi funzionali della molecola. I
legami sottoposti alle radiazioni assorbono energia che permette loro dei movimenti: l’assorbimento
di energia produce una variazione dell’intensitŕ della radiazione che viene
registrata poi dal detector. Ogni tipo di legame fra atomi e ogni gruppo funzionale ha un
assorbimento a una precisa lunghezza d’onda dell'infrarosso: quindi da tali
assorbimenti specifici si possono ricavare informazioni sui componenti della molecola e
quindi cercare di risalire ai componenti del campione. Gli atomi che fanno parte di un legame possono muoversi
in diversi modi: per esempio possono muoversi sul piano o fuori dal piano dando luogo a
differenti assorbimenti, inoltre i movimenti dei legami possono essere di vari tipi, per
esempio possono semplicemente, nel caso di due atomi legati ad uno centrale, avvicinarsi
ed allontanarsi, muoversi in due direzioni opposte fuori dal piano, ruotare su se stessi e
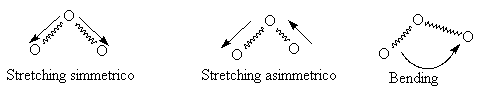
cosě via. In particolare si definiscono moti di stretching tutti
quelli che comportano la variazione della lunghezza del legame, e bending tutti i moti che
comportano la variazione dell’angolo di legame.
La scansione effettuata da uno spettrofotometro si
estende da 3800 a 200 cm-1. Lo spettro infrarosso si divide in tre zone
principali: la prima parte dello spettro (da 3800 a 1300 cm-1) detta zona dei
gruppi funzionali o vicino IR comprende le bande d’assorbimento dovute agli
stiramenti dei legami. Qui sono riconoscibili i legami X-H, i legami multipli C-Y, e le
deformazioni dei gruppi funzionali (Es NO2). La seconda parte di uno spettro IR (da 1300 a 650 cm-1)
č chiamata zona delle impronte digitali o "finger printing" perché mentre
nella prima zona si hanno assorbimenti da parte di tutte le sostanze, questa zona registra
assorbimenti dovuti alle vibrazioni totali di tutta la molecola (vibrazioni di
scheletro) e pertanto caratteristiche di ogni molecola. In questa porzione di spettro
č impossibile trovare due composti che abbiano lo stesso spettro, questa zona č appunto
detta delle impronte digitali poiché č caratteristica per ogni molecola. La terza ed ultima zona (da 650 a 200 cm-1)
detta zona del lontano IR comprende i legami di atomi pesanti, deformazioni di gruppi
privi d’idrogeno e vibrazioni di scheletro. Per l’eventuale riconoscimento di sostanze
incognite nei campioni ci si puň basare su tabelle che riportano le bande di assorbimento
dei gruppi funzionali e delle vibrazioni degli atomi oppure si confronta lo spettro in
esame con uno spettro di riferimento, in cui il campione non č altro che la presupposta
sostanza incognita pura. |
{kind=link}