|
|
Il retinoblastoma rappresenta la più comune neoplasia maligna intraoculare dell’infanzia. Nell’ambito di tutte le patologie oculari il retinoblastoma ha un’incidenza compresa tra lo 0,01% e lo 0,04% e risulta responsabile del 5% di tutti i casi di cecità. La sua incidenza varia, nelle differenti casistiche, da 1:14.000 a 1:34.000 nati vivi. La fascia di età più colpita è compresa tra la nascita e i 3 anni; l’età media alla diagnosi varia da 7 a 12 mesi nei casi bilaterali e da 18 a 24 mesi nei casi unilaterali. La neoplasia raramente si manifesta dopo i 3 anni. Meno del 2% dei casi sono diagnosticati dopo i 5 anni; tuttavia sono stati riportati casi di retinoblastoma anche nell’adulto. ASPETTI CLINICI Segni e sintomi I segni e sintomi del retinoblastoma dipendono dello stadio della malattia al momento della diagnosi e di solito sono riferiti dai genitori del bambino e/o dal pediatra. Il segno più comune è il ben noto riflesso bianco della pupilla (riflesso del gatto amaurotico): la leucocoria, presente nel 50-70% dei casi è dovuto alla crescita del tumore nel vitreo oppure ad un voluminoso focolaio nell’area papillo-maculare. La frequenza della leucocoria in ampie casistiche è variabile dal 32 al 78 %.
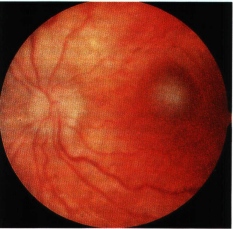
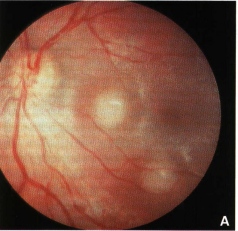
Secondo segno in ordine di frequenza è lo strabismo dovuto all’interessamento maculare con perdita della fissazione centrale e conseguente eso-exotropia. Si può manifestare isolato oppure associato alla leucocoria. Quando è isolato è dovuto a piccoli tumori maculari che interferiscono con la visione centrale. (Fig. 1).
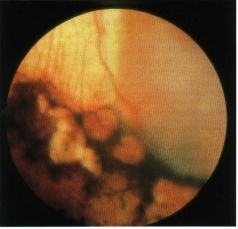
Non è raro scoprire, dopo un’accurata anamnesi clinica, che lo strabismo era comparso alcuni mesi prima della leucocoria. Meno frequentemente,il primo sintomo può essere un occhio rosso, dolente, con glaucoma secondario, presenza di pseudoipopion o ipoema . (Tabella II) Le modalità di crescita del retinoblastoma vengono ricondotte a 5 diversi tipi: endofitico, esofitico, misto endofitico e esofitico, infiltrante, diffuso e spontaneamente regredito. Le ultime 2 forme sono le più rare e sono descritte separatamente. La conoscenza delle modalità di crescita è indispensabile ai fini diagnostici, ma soprattutto per comprendere le differenze nella diffusione della neoplasia. Il retinoblastoma endofitico cresce dalla superficie interna della retina verso il vitreo. Pertanto oftalmoscopicamente si apprezza direttamente la massa neoplastica (bianco-giallastra) con i vasi retinici non più osservabili appena penetrato nel tumore. Questa forma ha crescita rapida, la massa è friabile e le cellule neoplastiche si disperdono nel vitreo (seeding vitreale) ove crescono come sferule biancastre simile a piccole palle di cotone (da non confondersi con aggregati infiammatori). Queste piccole raccolte neoplastiche possono depositarsi sulla retina e invaderla; questo insemenzamento retinico deve essere distinto, se possibile, da un tumore multicentrico (mutazione germinale): generalmente il seeding vitreale e i noduli neoplastici sulla superficie retinica e non entro la retina sono indicativi di insemenzamento neoplastico. Le cellule neoplastiche del vitreo possono diffondere verso il segmento anteriore depositandosi ovunque fino al quadro di uno pseudoipopion. Il retinoblastoma esofitico cresce dalla superficie esterna della retina verso la coroide, dapprima sollevando la retina e successivamente distaccandola. Pertanto i vasi retinici sono ben visibili sulla superficie neoplastica, che appare di colore più rosato rispetto alla forma endofitica. La proliferazione neoplastica è soprattutto indirizzata verso l’esterno: liquido sottoretinico, epitelio pigmentato retinico e coroide. Da qui l’infiltrazione lungo i nervi e i vasi ciliari può giungere all’orbita e alla congiuntiva dando luogo ad una mestatizzazione diffusa. I retinoblastomi di maggiori dimensioni sono di tipo misto, assumono le caratteristiche cliniche e di diffusione di entrambe le forme.
DIAGNOSTICA DIFFERENZIALE E’ talora difficile porre una diagnosi differenziale tra retinoblastoma ed una grande varietà di lesioni che lo possono simulare. (Tabella III) In 265 pazienti diagnosticati affetti da "pseudoretinoblastoma" il vitreo primario iperplastico e la fibroplasia retrolentale sono state le due condizioni più frequentemente simulanti sul retinoblastoma. Al Wills Eye Hospital di Philadelphia – Servizio di Oncologia Oculare – le lesioni che più frequentemente simulano un retinoblastoma sono: il vitreo primario iperplastico, la malattia di Coats e la toxocariasi oculare. La grande maggioranza delle lesioni riportate in tabella possono essere incluse usando un approccio clinico sistematico e moderne tecniche diagnostiche (anamnesi accurata, esame completo del segmento anteriore e posteriore dell’occhio, ecografia, TC, RM). La biopsia con ago sottile (FNAB) è potenzialmente pericolosa e dovrebbe essere evitata o al massimo riservata a casi accuratamente selezionati.
La diagnosi di retinoblastoma è quindi basata sui dati clinici ed oftalmoscopici e le indagini invasive sono raramente necessarie. Si raccomanda, comunque, prima di adottare una qualsiasi procedura diagnostica invasiva di consultare un oncologo oculare. Di fondamentale importanza è un’accurata anamnesi; talora una storia familiare di retinoblastoma non viene riferita perché i genitori ignorano la natura ereditaria della malattia; la perdita di un occhio nell’infanzia può essere giustificata, dai genitori al bambino, come il risultato di un trauma e non come la vera ragione dell’enucleazione. Un’anamnesi che riveli prematurità, infezioni neonatali o la presenza di anomalie sistemiche (dismorfimi, ritardo mentale, segni cutanei di facomatosi) richiede un’attenta valutazione ed adeguati protocolli dignostici. L’esame del FUNDUS dei parenti di primo grado può spesso documentare la presenza di un retinoma o di un retinoblastoma spontaneamente regredito e quindi "smascherare" il carattere ereditario del tumore. Uno studio completo comprende: un esame del segmento anteriore e posteriore di entrambi gli occhi, una valutazione pediatrica e radiologica e studi genetici (Tabella IV).
L’esame di un occhio con sospetto retinoblastoma va sempre seguito in anestesia generale e va rispettato un protocollo di procedure clinico-diagnostiche (Tabella V).
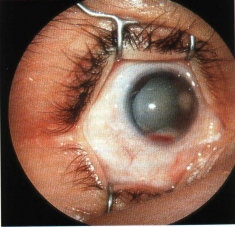
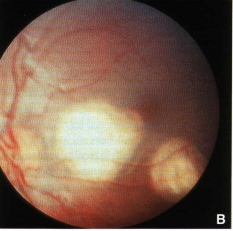
L’oftalmoscopia binoculare indiretta deve essere scrupolosa, con indentazione sclerale per l’individuazione di eventuali piccoli focolai tumorali in estrema periferia retinica. Il disegno del FUNDUS e la documentazione fotografica sono di estrema importanza clinica e scientifica per il follow-up del piccolo paziente. Sul disegno del fondo vanno riportati tutti i focolai tumorali e le rispettive dimensioni ed altre eventuali annotazione (presenza di distacco di retina, emorragie). L’esame del segmento anteriore deve mirare alla ricerca di segni di disseminazione iridea o in camera anteriore: neovascolarizzazione, pseudoipopion e ipoema (Fig. 2).
Il controllo della pressione intraoculare è importante in quanto non è raro un glaucoma secondario in casi di retinoblastomi voluminosi ed avanzati. La diagnosi ecografica di retinoblastoma, viene effettuata con la metodica B- e A-Scan standardizzata. Il quadro ecografico tipico è stato ampiamente descritto in letteratura ed è rappresentato dal retinoblastoma endofitico con calcificazioni. In B-Scan, il retinoblstoma si presenta come una lesione solida, mono o plurilobata, rilevata sul piano retinico, con trama acustica piuttosto densa ed omogeneità variabile che determina attenuazione degli ultrasuoni con ombreggiamento degli echi provenienti dal tessuto orbitarlo che aumenta in proporzioni alla presenza di calcificazioni. Accanto ai focolai, è possibile repertare strie esogene da distacco di retina satellite; l’impegno vitreale è visibile se vi è seeding o dopo un eventuale trattamento fotocoagulativo per una coartazione del vitro iuxtalesionale. Con la tecnica A-scan standardizzata, il tracciato presenta un picco di aperture ad alta reflettività, ma quasi mai massimale, in quanto la superficie retinica appare disgregata dalla crescita tumorale; la struttura interna è piuttosto irregolare, con reflettività alta e picchi sovramassimali corrispondenti alle calcificazioni. L’attenuazione degli ultrasuoni si evidenzia con una rapida e progressiva riduzione di altezza degli echi orbitari che decrescono dietro il picco sclerale. I margini posteriori della lesione sono evidenziabili dall’esame del picco sclerale ad alta reflettività. Quando la sclera è integra, il picco sclerale è ben evidente dietro il tessuto patologico. L’invasione sclerale comporta una riduzione della reflettività del picco sclerale di chiusura che si confonderà con gli echi retinici del tumore. Nel retinoblastoma a crescita esofitica, soprattutto negli stadi iniziali, la diagnosi è più complessa con entrambe le metodiche, in quanto la lesione appare poco rilevata sul piano retinico e si confonde con la porzione ad alta ecogeneicità della sclera. Le forme non calcificate, descritte come diffuse, si manifestano sia in B-che in A-scan standardizzato con un impegno vitreale diffuso, senza masse solide o aree necrotiche, ma con ispessimento irregolare della retina. Dai differenti quadri descritti, emerge che la variabilità di crescita endofitica ed esofitica, la multifocalità delle lesioni, la presenza o meno di calcificazioni giustificano una certa variabilità del quadro ecografico, sia in B-scan che in A-scan standardizzato, la cui interpretazione non può prescindere dalla conoscenza dei quadri clinici e dalla esperienza dell’esaminatore. La TC mostra, di solito, la presenza di una lesione intraoculare di dimensioni variabili, calcifica nel 95% dei casi , a sviluppo endofitico, diffuso o misto. La massa è iperdensa rispetto al vitreo; dopo iniezione di mezzo di contrasto è apprezzabile un incremento tomodensitometrico variabile,da modesto a marcato. La TC spesso non è in grado di differenziare il tumore dall’essudato sottoretinico e non è sensibile nella valutazione di un’eventuale estensione al nervo ottico: può essere sufficiente per escludere un interessamento intracranico . La RM dimostra generalmente una massa isointensa o moderatamente iperintensa rispetto al vitreo nelle sequenze a TR breve e più o meno marcatamente ipointensa nelle sequenze a TR lungo. Queste ultime sono in grado di differenziare il tumore dall’essudato sottoretinico (iperintenso). Tale valutazione è possibile anche nelle sequenze a TR breve, dopo iniezione di mezzo di contrasto paramagnetico, qualora la masserella presenti un sufficiente potenziamento. Nonostante La TC resti l’esame più sensibile per rilevare le calcificazioni, le sequenze RM "gradient echo" consentono, nella maggior parte dei casi, di differenziare le aree calcifiche che appaiono nettamente ipointense con un decadimento di segnale maggiore rispetto alle sequenze "spin echo". La RM ha inoltre una sensibilità superiore nello studio dei nervi ottici e del versate intracranico. Ritardo di diagnosi Indiscutibilmente un ritardo nella diagnosi comporta uno stadio più avanzato della malattia ed una prognosi peggiore. Spesso fattori sociali e culturali possono influenzare il momento della diagnosi e della terapia. I seguenti motivi possono essere alla base di un ritardo diagnostico:
Manifestazioni insolite, retinoblastoma infiltrante diffuso e retinoma Talvolta il retinoblastoma può manifestarsi con segni e sintomi atipici che possono, ad una valutazione non esperta, fuorviare la diagnosi. Tra i segni "insoliti" più frequenti troviamo quelli di tipo infiammatorio: cheratocongiutivite o pseudouveite con ipopion, endoftalmite (fino a casi estremi di tisi del bulbo) e cellulite orbitarla. Tali segni"inusuali" sono più frequentemente presenti nei casi di retinoblastoma infiltrante diffuso. Quest’ultimo costituisce una rara forma clinica (pari al 2% circa) che si manifesta tardivamente (età media di diagnosi: 6 anni), predilige il sesso maschile, è unilaterale e presenta i segni tipici di una pseudouveite. La neoplasia cresce infiltrando la retina senza formare masse visibili, le calcificazioni sono molto scarse o assenti. L’invasione della camera anteriore provoca uno pseudoipopion contenente cellule tumorali poco adese tra loro, tanto da cambiare facilmente di forma, modificando la posizione della testa e del bulbo. L’occhio non mostra segni di congestione vascolare pericheratica o episclerale e "l’infiammazione" non risponde al trattamento antinfiammatorio. Una paracentesi diagnostica della camera anteriore in questi casi, anche se ripetuta, può dare risultati negativi (Tabella VI).
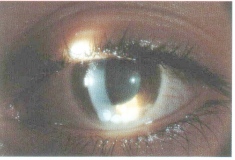
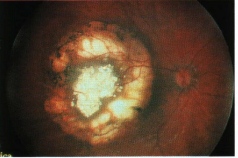
Altri segni "inusuali", ma tipici di uno stadio avanzato della malattia, sono: eterocromia e rubeosi iridea nel quadro di un glaucoma secondario. Tale stadio è un’indicazione all’enucleazione, anche in caso di dubbio diagnostico, per la presenza di dolore bulbare intrattabile e la totale assenza di capacità visiva dell’occhio interessato. In fine un distacco retinico molto esteso, associato ad elevata quantità di liquido sottoretinico, può provocare difficoltà nel riconoscere un focolaio tumorale e porre problemi di diagnosi differenziale soprattutto verso la malattia di Coast, in tal caso sono di assoluto ausilio le tecniche ecografiche e neuroradiologiche. Si parla di retinoblastoma tardivo quando la diagnosi viene posta dopo i 5 anni di età. L’incidenza di tali forme è piuttosto modesta: si aggira intorno al 2% se si considerano solo i veri retinoblastomi tardivi e raggiungere l’8,5% secondo Shields se si comprendono anche le forme benigne di retinoma. Si tratta di retinoblastomi unilaterali, sporadici, che possono manifestarsi con le stesse caratteristiche cliniche del retinoblastoma precoce, quali leucocoria e strabismo, ma più frequentemente presentano dei sintomi atipici che possono provocare un ritardo di diagnosi, con grave compromissione per la prognosi. I segni atipici che più frequentemente accompagnano un retinoblastoma tardivo sono: ipopion, ipoema, uveite, endoftalmite, emorragia vitreale e cellulite orbitaria (Fig. 3). La presenza di dolore e diminuzione del visus, sintomi non valutabili nel retinoblastoma precoce, costituiscono un altro elemento importante nella diagnosi differenziale (Tabella VII). Il retinoblastoma tardivo deve quindi essere considerato nella diagnosi differenziale di ogni bambino che presenti una massa intraoculare, o segni di infiammazione o di emorragia intraoculare. Un ritardo di diagnosi può compromettere la prognosi, non solo al fine di un trattamento conservativo, ma anche quoad vitam, in quanto tali forme presentano un’aggressività clinica che non è mitigata dall’insorgenza tardiva.
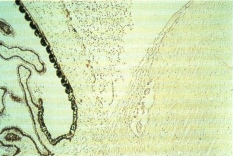
L’incidenza del glaucoma secondario nel retinoblastoma è compresa, nelle varie casistiche, tra il 2% e il 23% il glaucoma secondario può manifestarsi con una sintomatologia eclatante, caratterizzata da arrossamento oculare, dolore, edema corneale, buftalmo, ma in alcuni solo un attento esame biomicroscopico del segmento anteriore e la misurazione della pressione intraoculare consentono l’identificazione del glaucoma. Il glaucoma secondario è indice di uno stadio avanzato della malattia e rappresenta pertanto un elemento prognostico sfavorevole ed una chiara indicazione all’enucleazione. Vari studi hanno focalizzato l’attenzione sui meccanismi alla base della perturbazione idrodinamica responsabile dell’ipertono oculare. Questi meccanismi possono essere isolati o agire in combinazione e sono costituiti da: blocco angolare per chiusura dell’angolo irido-corneale in seguito all’avanzamento del diaframma irido-lenticolare dovuto al dislocamento anteriore del vitreo sotto la spinta della massa tumorale; blocco del trabecolato, per la presenza di mocrofagi in vario stadio di degenerazione, di cellule tumorali e di detriti cellulari che ostacolano il deflusso dell’umore acqueo (Fig. 4); neovascolarizzazione dell’iride ad opera di neovasi che di solito appaiono inizialmente sul margine pupillare e, procedendo sulla superficie dell’iride, raggiungono l’angolo irido-corneale ed il trabecolato provocandone l’ostruzione fibrovascolere. I reperti istopatologici hanno evidenziato la prevalenza del meccanismo patogenetico combinato rispetto a quello isolato; il blocco trabecolare associato alla neovascolarizzazione iridea è risultata essere la combinazione più frequente.
RETINOMA E RETINOBLASTOMA SPONTANEAMENTE REGREDITO Il termine retinoma (o retinocitoma) è usato per definire un tumore benigno di origine retinocitica. Brenda Galleie ha suggerito che queste lezioni, definite in passato come retinoblastomi spontaneamente regrediti, sono in realtà tumori retinici benigni. I retinomi sono istologicamente composti da cellule apparentemente benigne senza evidenza di negrosi o di attività mitotica, ma con numerose fleurettes; i retinomi hanno almeno due delle tre caratteristiche elencate in tabella VIII. La trasformazione maligna di un retinoma è molto rara. Nella nostra esperienza adulta con retinoma ha sviluppato dopo due anni di follow-up un retinoblastoma.
Osservazioni simili sono riportate in letteratura da Shields e da Abramson (Fig. 5).
Il termine retinoblastoma spontaneamente regredito deve essere riservato per descrivere occhi in tisi con calcificazioni al loro interno. Nella regressione spontanea del retinoblastoma sono stati implicati: riduzione del flusso ematico; necrosi tumorale; "calcio-inibizione" della crescita tumorale e meccanismi di risposta immunitaria dell’ospite. RETINOBLASTOMA ASSOCIATO A DELEZIONE DEL CROMOSOMA 13 (13q DELETION SYNDROME) E GENETICA DEL RETINOBLASTOMA In circa il 5% dei casi il retinoblastoma può essere associato ad alterazioni cromosomiche. In tal caso si accompagna di solito ad anomalie sistemiche ed oculari. In letteratura sono descritti diversi tipi di aberrazione cromosomica in associazione al retinoblastoma: trisonia 13, traslocazione 13:X, trisomia 21, trisomia X-21 e sindrome di Klinefelter. in tutte queste associazioni il numero di casi descritto è così piccolo da non permettere una reale valutazione delle correlazioni esistenti tra la sindrome stessa e il retinoblastoma. L’associazione del retinoblatoma con delezione del braccio lungo di un cromosoma del gruppo D (cromosoma 13, 14 e 15) è stata descritta sin dal 1963. Successivamente, sono state individuate due specifiche sindromi denominate rispettivamente "Sindrome del cromosoma D ad anello" e "Sindrome da delezione del cromosoma 13". La delezione del cromosoma 13 comporta la presenza di anomalie congenite, sistemiche e oftalmologighe che difficilmente passano inosservate (Tabella IX). Le anomalie oftalmologiche sono costituite da: ipertelorismo, epicanto, ptosi, coloboma ueale, microftalmo, rima palpebrale antimongoloide, ipoplasia del nervo ottico. Le alterazioni sistemiche sono ancora più evidenti e dovrebbero sempre far sospettare una delezione del cromosoma 13, quando presenti in associazione tra loro. Ricordiamo tra queste: ritardo psicomotorio, microcefalia, padiglioni auricolari malformati, ruotati ed abbassati, ipoplasia o assenza del pollice che appare inserito più prossimamente all’arto superiore, naso a "sella" con punta arrotondata, incisivi superiori prominenti, collo corto con pliche laterali, bozze frontali prominenti, frenulo labiale allungato, ano imperforato, malformazioni cardiache e scheletriche. La presenza di anomalie sistemiche ed oculari che facciano intuire aberrazioni cromosomiche a carico del cromosoma 13 dovrebbero sempre far sospettare l’associazione con retinoblastoma e quindi condurre ad una diagnosi precoce della neoplasia.
Tale associazione è descritta in letteratura in percentuale variabile a seconda dei vari Autori: dal 2,8% (Pratt) al 7% (Shields). La nostra esperienza clinica ha documentato tale associazione nel 4% dei casi di retinoblastoma. In linea generale esiste in letteratura la tendenza a considerare per tale tipo di retinoblastoma un comportamento clinico meno aggressivo rispetto al tumore non associato a delezione del cromosoma 13. L’esperienza personale non concorda con queste osservazioni, si è osservata, viceversa, una molteplicità di aspetti clinici, che comprendono anche casi con metastasi e secondi tumori. L’associazione del retinoblastoma con delezione del cromosoma 13 è inoltre descritta quasi sempre come un caso sporadico, molto raramente come caso di delezione familiare. Secondo l’ipotesi genetica corrente, tutte le forme di retinoblastoma sono l’effetto di un’alterazione del gene RB1. Il locus del gene RB1 è situato nella regione 13q14.1 – 14.2, ossia nella porzione prossimale del braccio lungo del cromosoma 13. Il gene ha una lunghezza di circa 180 Kilobasi (Kb) ovvero 180.000 basi nucleotidiche e codifica un prodotto di circa 4,7 Kb (ciò si verifica in quanto ogni gene è costituito di un insieme di porzioni che vengono trascritte in RNA messaggero, definite esoni e porzioni che non vengono trascritte, definite introni. Queste ultime sono, in genere, molto ampie rispetto alle prime. Solo le porzioni codificanti vengono trascritte in RNA messaggero, mentre, per effetto del processo chimico definito "splicing", tutte le porzioni non codificanti vengono eliminate. Questo è, in sintesi, il processo attraverso il quale da un gene molto grande si può ottenere un trascritto piuttosto piccolo). Il gene RB1 è stato caratterizzato come "tumor suppressor ( o suppresser) gene", ossia un gene che, in condizioni normali, tiene sotto controllo la capacità proliferativa della cellula. Di fatto, il prodotto normale del gene RB1 si lega al DNA e controlla il ciclo cellulare nella fase di transizione da G1a S. Dal punto di vista della funzione e del tipo di gene coinvolto nella genesi del retinoblastoma, è importante sottolineare come la malattia si sviluppi soltanto quando entrambe le copie del gene perdono la loro funzione. E’ per tale motivo che il gene RB1, come tutti gli altri "tumor suppressor genes" viene anche definito gene "recessivo" per il cancro. Questa definizione, tuttavia, può indurre ad equivoco ove si consideri la natura ereditaria del retinoblastoma. Infatti nella forma ereditaria della malattia, un cromosoma contenente la forma mutante del gene RB1, viene trasmesso alla progenie con le modalità tipiche dell’ereditarietà autosomica dominante di tipo mendeliano. In questo caso, tutte le cellule dell’organismo che ha ereditato il gene mutato possiedono una sola copia normale del gene in questione. Il retinoblastoma si svilupperà quando questa copia normale sarà inattivata o perduta mediante il processo definito "perdita di eterozigoti" (LOH = Loss of Heterozygosity, degli Autori anglosassoni). L’ipotesi secondo cui alla base del retinoblastoma vi è un processo a due tappe (in- attivazione funzionale consecutiva di due copie dello stesso gene), era stata avanzata già molto tempo prima che il gene RB1 fosse stato individuato e caratterizzato. Tale ipotesi si era resa necessaria per spiegare almeno alcuni degli aspetti clinici/epidemiologici fondamentali della malattia. E’ infatti noto che, in generale, è possibile distinguere due forme di retinoblastoma: la forma ereditaria (responsabile di circa il 40% dei casi), in cui la prima mutazione a carico del gene RB1 è già presente nella linea germinale e la forma sporadica (responsabile del restante 60% dei casi), in cui sia la prima che la seconda mutazione avvengono a carico della cellula somatica (retinoblasto). L’ipotesi che prevedeva il succedersi di due eventi mutageni a carico di un gene chiave ( poi indicato come RB1 e completamente caratterizzato dal punto di vista funzionale) sembrava spiegare bene la realtà clinica. In sostanza si ammetteva che, nel caso in cui il primo evento fosse a carico della cellula germinale (retinoblastoma ereditario), tutte le cellule somatiche del feto ne risultassero colpite, con conseguente aumento delle probabilità che il secondo evento si potesse verificare e non solo a carico dei retinoblasti. Si riteneva di spiegare in tal modo la maggiore precocità dei retinoblastomi ereditari, come pure il più frequente interessamento di entrambi gli occhi e la maggior tendenza, propria di questi soggetti, a sviluppare un secondo tumore non oculare (più spesso un osteosarcoma). Al contrario, nel caso in cui sia il primo che il secondo evento si verificassero a carico del retinoblasto, senza coinvolgimento delle cellule germinali, si riteneva di poter spiegare perché, nel retinoblastoma sporadico vi fosse una maggior tendenza all’interessamento di un solo occhio, l’età di insorgenza fosse più tardiva e non vi fosse tendenza a sviluppare un secondo tumore non oculare. Molto di quanto originariamente ipotizzato è stato confermato dai successivi studi di genetica molecolare che, tra l’altro, hanno condotto alla scoperta di RB1 e di altri geni soppressori. Molto, tuttavia, resta ancore da fare per comprendere a fondo l’eziopatogenesi della malattia e, sebbene l’onda emotiva della scoperta dei geni soppressori non sia ancora completamente esaurita, la complessa realtà clinico/epidemiologica ed i primi dati importanti sugli screening genetici per il retinoblastoma, stanno lentamente ma inevitabilmente conducendo verso la ricerca di plausibili alternative all’ipotesi del doppio evento. Nel giudizio negativo sull’ipotesi delle due tappe sembrano pesare i risultati delle indagini molecolari che avrebbero dovuto confermare l’ipotesi stessa. E’ infatti noto che alterazioni del gene RB1 si trovano in una percentuale di casi variabile dal 20 all’80% dei soggetti affetti, mentre le stesse alterazioni del gene RB1 si troverebbero in un grande numero di altre neoplasie (polmone, mammella, utero, vescica, colon-retto, prostata e fegato, solo per menzionare alcune tra le più rilevanti). Ciò implica che non solo le alterazioni di RB non rappresentano un indicatore sufficientemente sensibile e specifico del retinoblastoma, ma che lo stesso nesso di casualità ipotizzato, tra alterazioni di RB1 e retinoblastoma, non trova giustificazione nei dati fino ad oggi disponibili. D’altra parte, sempre per quanto riguarda i dati relativi alla genetica molecolare, alcuni importanti studi su grandi casistiche hanno ampiamente dimostrato che:
Globalmente, è possibile affermare che l’ipotesi genetica non risulta confermata dai dati epidemiologici e molecolari a nostra disposizione e che, allo astato attuale delle conoscenze, la biologia molecolare, pur disponendo di numerose tecniche di analisi del gene RB1, ancorché chiarire alcuni fondamentali aspetti eziopatogenetici del retinoblastoma, abbia aggiunto notevoli complicazioni. I primi tentativi di diagnosi prenatale mediante tecniche di biologia molecolare, utilizzavano la "linkage analysis", un metodo molto indiretto che doveva consentire la costruzione della mappa del gene malato nel tumore e la sua identificazione nel genotipo costituzionale dell’individuo affetto. Senza inoltrarci in inutili dettagli tecnici, è possibile affermare che tale tecnica, anche nella sua variante "intragenica" subentrata dopo la pubblicazione della sequenza intera del gene RB1, è stata rapidamente abbandonata. Alla "linkage analysis" sono succedute indagini più raffinate basate sul comportamento del DNA denaturato quando sottoposto ad elettroforesi. Queste tecniche, unitariamente indicate con il termine di "analisi conformazionale" del DNA presentano un gran numero di varianti tutte più o meno rivolte ad aumentarne la sensibilità. In linea di principio, mentre il DNA a doppia elica, sottoposto ad elettroforesi, migra con una velocità inversamente proporzionale alla sua grandezza, il DNA denaturato (con calore o con agenti chimici) migra in rapporto alla conformazione che il singolo filamento assume dopo denaturazione, essendo, tale configurazione, strettamente condizionata dalla sequenza nucleotidica. Accade pertanto che, mentre con l’elettroforesi applicata al DNA a doppia elica non sia possibile distinguere due diversi DNA che abbiano lo stesso numero di basi, con l’analisi conformazionale ciò diventa possibile in quanto due filamenti di pari lunghezza ma di sequenza diversa, migrano, nel campo elettroforetico, con velocità diverse. Quindi, assumendo che la sequenza delle porzioni codificanti di un gene (esoni) sia identica per tutti gli individui di una specie, minime variazioni nella sequenza (mutazioni puntiformi), possono essere facilmente identificate mediante l’analisi conformazionale. L’analisi conformazionale del DNA assume dunque grande rilevanza nello screenig delle mutazioni per l’individuazione delle porzioni mutate di un gene. Le porzioni "alterate" vengono successivamente sottoposte a sequenziamento diretto al fine di caratterizzare qualitativamente l’alterazione in causa nella malattia. La più nota ed applicata, tra le tecniche di analisi conformazionale del DNA nel retinoblastoma, è la cosiddetta "Single Strand Conformation Polymorphism analysis" o SSCP, ma, come anticipato, numerose varianti di questa tecnica sono riportate in letteratura e, tra queste, la DGGE (Denaturing Gradient Gel Elettrophoresis), la TGGE (Temperature Gradient Gel Elettrophoresis), la CDGE (Consant Denatrant Gradient Gel Elettrophoresis) ed altre ancora. Il panorama delle tecniche di biologia molecolare, applicate allo studio delle mutazioni del gene RB1, è, ovviamente, di gran lunga più ampio e complesso di quanto riportato. Tecniche per così dire "alternative" e più "economiche" rispetto a quelle di analisi conformazionale, quali ad esempio il sequenziamento diretto del gene o l’elettroforesi bidimensionale, sono state recentemente proposte da alcuni Autori. Qui ci limiteremo a sottolineare che la maggiore "economicità" delle tecniche proposte è, per il momento, solo presunta: il sequenziamento diretto si presta a molte critiche di carattere tecnico ove proposto per l’impiego come test di "screening" e l’elettroforesi bidimensionale è una tecnica non ancora standardizzata per la diagnostica del retinoblastoma e non se ne conosce la reale sensibilità. Nella nostra esperienza e con il conforto di una cospicua parte della letteratura specializzata possiamo concludere affermando che la SSCP, ove applicata secondo criteri procedurali rigorosi e standardizzati, resta la tecnica di elezione per la diagnostica di base nelle mutazioni del gene RB1, mentre il sequenziamento va riservato all’analisi qualitativa delle sequenze che, con la SSCP, mostrano alterazioni dei modelli di migrazione. ISTOPATOLOGIA Il retinoblastoma, tumore che origina dal neuroepitelio retinico, è caratterizzato dalla presenza di aggregati cellulari in forma sferica, le rosette, in cui le cellule neoplastiche si dispongono con un prolungamento cellulare rivolto verso l’interno. Ritenute rappresentare espressione di differenziazione sono in realtà cellule maligne e si possono distinguere in: rosette di Homerwright (caratteristiche del retinoblastoma; si possono osservare anche nel pinealoblastoma e nel medulloblastoma) e rosette di Flexner-Winterstainer, in cui il prolungamento citoplasmatico delimita uno spazio centrale otticamente vuoto. Ulteriore espressione di differenziazione è rappresentato dalle fleurettes, in cui l’appendice citoplasmatica è rivolta all’esterno. Nel contesto del tumore sono sempre presenti aree di necrosi coagulativa, in quantità variabile. In genere le cellule neoplastiche sopravvivono nelle aree più prossime ai vasi (per evidente necessità di ossigeno) formando le cosiddette "pseudorosette". Nelle aree necrotiche si depositano sali di calcio, che danno origine alle calcificazioni, mentre detriti molecolari delle cellule necrotiche si possono depositare nelle pareti vascolari a causare vasofilia perivascolare. Nella stadiazione del retinoblastoma post-enucleazione il ruolo dell’anatomopatologo è di rilevante importanza in quanto deve essere sempre valutata e quantificata accuratamente l’infiltrazione delle seguenti strutture: coroide, sclera (iniziale, focale, a tutto spessore, diffusa), corpo ciliare, camera anteriore ed iride; ma soprattutto l’infiltrazione del nervo ottico: assente, limitata alla porzione prelaminare, estesa alla laminare o oltre la lamina cribrosa: prima o a livello del piano di sezione chirurgica del nervo ottico. Questi elementi condizioneranno il piano terapeutico generale. E’ necessario inoltre ricordare che l’anatomopatologo deve essere sempre informato preventivamente dell’invio di un bulbo oculare presumibilmente affetto da retinoblastoma affinché possa predisporre le adeguate indagini su tessuto fresco: il bulbo va inviato a fresco e mai già fissato in formalina. La maggior parte dei retinoblastomi mostra le caratteristiche modalità di crescita rapida ed invasiva dei tumori neuroblastici dell’infanzia. La metastatizzazione avviene secondo quattro modalità: infiltrazione diretta (dal nervo ottico sino all’encefalo e/o dalla coroide direttamente attraverso i canali sclerali o dopo l’invasione sclerale massiva, i tessuti molli orbitali e da qui all’osso), dispersione ( alle leptomeningi e di qui alla circolazione liquorale e all’encefalo), disseminazione ematogena (possibile dopo l’invasione extraoculare; diretta a polmoni, ossa, cervello e altri organi) e linfatica (dopo l’invasione del segmento anteriore attraverso i linfatici della congiuntiva e delle palpebre; non vi sono vasi linfatici nell’orbita). TRATTAMENTO Il trattamento del retinoblastoma, che comprende diverse opzioni terapeutiche, può essere frutto solo di un’attenta e completa valutazione del singolo caso clinico e del raffronto del caso con le stadiazioni attualmente in uso. La scelta terapeutica può essere affidata solo a persone esperte di oncologia oculare, perché anche la procedura apparentemente più semplice, quale l’enucleazione, richiede particolari attenzioni tecniche e le più moderne terapie oncologiche integrate richiedono la collaborazione abituale con oncologi (pediatri) e radioterapisti. In base alla stadiazione clinica sono possibili due approcci fondamentali: enucleazione o terapia conservativa. L’enucleazione è stata per decenni la terapia più efficace ed è tuttora nei casi indicati di seguito la più valida scelta terapeutica. Questo intervento ha permesso di salvare numerosissime vite umane, trasformando il retinoblastoma da tumore diffusivo e incurabile a malattia curabile. La frequenza di enucleazione è diminuita negli ultimi anni grazie a valide terapie alternative, le cui indicazioni sono riportate più avanti. Le terapie conservative sono le seguenti: fotocoagulazione, crioterapia, radioterapia con placche episclerali, chemioriduzione e termoterapia, radioterapia da sorgente esterna (teleterapia). L’enucleazione ha oggi le seguenti indicazioni:
La richiesta dei genitori di non enucleare il bulbo affetto da retinoblastoma per cui vi sia indicazione all’enucleazione va attentamente valutata ricordando ai genitori stessi i rischi per la sopravvivenza del bambino di una diffusione extraoculare della malattia. La scelta terapeutica iniziale dipende da una serie di fattori quali: l’uni- o multifocalità, la sede e le dimensioni del tumore; il seeding vitreale diffuso o focale l’età del bambino. Un corretto approccio terapeutico iniziale richiede, come già sottolineato, un’accurata stadiazione della malattia. La stadiazione più comunemente in uso è quella di Reese ed Ellsworth ed è basata sulla valutazione oftalmoscopica dell’estensione tumorale (Tab. X ). Altri sistemi di classificazione proposti sono quelli di: Pratt (basato sulla estensione intraoculare del retinoblastoma), Standard (basata sui dati epidemiologici) e la classificazione di Essen. L’International Committee per lo studio del retinoblastoma sta elaborando un nuovo sistema di stadiazione basato fondamentalmente sulla sede, dimensioni e uni- o multifocalità della malattia. La fotocoagulazione del retinoblastoma ebbe inizio nel 1955 quando Gerd Meyer-Schwickerth usò la fotocoagulazione allo Xenon per trattare un retinoblastoma. Secondo la tecnica suggerita dal pioniere di questa terapia, il tumore viene inizialmente circondato da un anello di "spots" su retina sana (tecnica indiretta) e poi viene trattato direttamente sulla sua superficie (tecnica diretta). La fotocoagulazione con Xenon è stata usata per decenni con enorme successo, successivamente sostituita da quella con laser. Quest’ultima non ha però rappresentato un alternativa sempre sicura ed efficace, infatti diverse sono state le indicazioni e le limitazioni al suo uso. Recentemente l’introduzione della termoterapia transpupillare (vedi anche il paragrafo sulla terapia del melanoma coroidale) ha rivalutato l’uso delle sorgenti laser associate alla chemioterapia. Le complicanze più comuni della fotocoagulazione con Xenon comprendono: occasionali emorragie retiniche, trazioni retiniche, danno irideo (specialmente in casi di modesta midriasi), edema corneale e opacità del cristallino. I quadri di regressione osservati dopo fotocoagulazione dipendono dalle dimensioni, in particolare dallo spessore del tumore. I tumori piccoli appaiono come una cicatrice avascolare piatta atrofica a bordi pigmentati; tumori più grandi presentano una marcata coartazione, una ridotta vascolarizzazione ed un aspetto traslucido molto simile a quello definito "fish-flesh". La vascolarizzazione del tumore influisce sulla sensibilità alla fotocoagulazione: più vascolarizzato è il focolaio tumorale, migliore sembra essere la risposta al trattamento. La fotocoagulazione è controindicata nei tumori adiacenti al nervo ottico e quando vi è seeding vitreale. Oggi la fotocoagulazione come trattamento di prima scelta o dopo chemioriduzione è riservata a focolai del polo posteriore e della media periferia retinica, risulta di più difficile esecuzione nei tumori di estrema periferia retinica ove necessita un’energica indentazione sclerale. Le energie da usare sono "personalizzate" per ogni caso e comunque variano in base allo spessore e alla sede del focolaio tumorale (Fig. 6).
La criocoagulazione è indicata per tumori piccoli anteriori all’equatore e vicini all’ora serrata; questi spesso sono i cosiddetti "nuovi tumori" cioè focolai comparsi più tardivamente, durante il trattamento e follow-up dei focolai del polo posteriore e della media periferica; a volte "nuovi" tumori compaiono dopo il trattamento radiante.
Nei più recenti protocolli conservativi del trattamento del retinoblastoma la criocoagulazione trova una nuova, importante collocazione in associazione alla monochemioterapia con il carboplatino: sembra che la rottura della barriera ematoretinica prodotta dalla criocoagulazione permetta una diffusione significativamente superiore del farmaco nel vitreo con conseguenti ottimi risultati nel trattamento del seeding vitreale. La crioterapia è efficace nei tomori fino a 3-4 mm di diametro e 2 mm di spessore, spesso sono necessarie sedute ripetute di trattamento. Il tumore deve essere perfettamente localizzato. con massima indentazione con la sonda da criocoagulazione mediante oftalmoscopia indiretta e viene criocoagulato con tre successivi spots (nella stessa sede) a temperatura di –80° C, di 30-60 sec. di durata ciascuno. La più frequente complicanza è l’edema congiuntivale e palpebrale. L’uso di placche episclerali radioattive (Co 60, Ir 192, Ru 106, I 125) come trattamento di prima scelta del retinoblastoma è riservato alle seguenti condizioni:
Come trattamento di seconda scelta, le placche vengono utilizzate nei tumori nuovi e recidivanti, ove la crioterapia e/o il laser in combinazione con la chemioterapia o da soli non hanno dato il risultato desiderato. Shields raccomanda l’uso della placca radiante nel retinoblastoma per lo più solitario di 4-12 mm di diametro e in tutti i retinoblastomi che manifestano un seeding vitreale focale dato che le radiazioni sono capaci di distruggere le cellule neoplastiche a livello della retina e del vitreo sovrastante. Gli isotopi attualmente impiegati sono il Rutenio 106 e lo Iodio 125. La brachiterapia (placche episclerali) non espone l’area peribulbare a significative quote di radiazione ionizzante. Le principali complicanze sono: la corioretinopatia da radiazioni (rara date le basse dosi impiegate) e le opacità del cristallino (dipendenti dalla effettiva dosa di irradiazione al cristallino stesso). La chemioterapia, impiegata in passato solo nel retinoblastoma extraoculare e metastatico, è attualmente divenuta un trattamento di prima scelta nel retinoblastoma intraoculare. I vantaggi dell’utilizzo della chemioterapia sistemica sono i seguenti: riduzione delle dimensioni dei focolai da trattare e quindi possibilità di utilizzare minori energie termiche; controllo delle micrometastasi durante il trattamento conservativo. Tra i farmaci utilizzati nella chemioterapia del retinoblastoma la combinazione di carboplatino ed etoposide, già in uso nel trattamento del neuroblastoma, ha dato ottimi risultati. L’esperienza accumulata dimostra che la riduzione delle dimensioni dei focolai intraoculari indotta dalla chemioterapia aumenta l’efficacia delle modalità locali di trattamento, sia per effetto della chemioriduzione del bersaglio, sia per effetto del sinergismo tra chemioterapia e coagulazione. Più dettagliatamente, la chemioterapia è indicata nelle seguenti condizioni:
La chemioterapia come trattamento di prima scelta, è inutile in occhi con glaucoma secondario, dove l’unica indicazione è l’enucleazione. in alcuni casi con diffuso seeding vitreale, la chemioterapia può avere successo in associazione alla criocoagulazione; sembra infatti che la criocoagulazione incrementi fini a trenta volte la quantità di carboplatino che arriva nella cavità vitreale. La risposta dei focolai tumorali alla chemioterapia è imprevedibile e a volte può sconvolgere radicalmente eventuali scelte terapeutiche programmate alla prima osservazione. Non è raro osservare la coartazione e la trasformazione calcifica di grossi focolai tumorali, ma anche la scomparsa del distacco retinico satellite. I quadri di regressione osservati dopo chemioterapia, si possono così riassumere:
Il numero dei cicli di chemioterapia viene programmato sulla base delle dimensioni dei focolai (in particolare sullo spessore) e del loro numero. I controlli vengono effettuati ogni 21-28 giorni e cioè alla fine di ogni ciclo di chemioterapia; durante il controllo oftalmoscopico in anestesia generale, viene decisa, di volta in volta, la necessità di trattamenti crio- e/o fotocoagulativi. La termo-chemioterapia (TCT) è una metodica introdotta recentemente che associa la termoterapia transpupillare con la monochemioterapia con carboplatino, potenziando gli effetti di entrambe. La TCT è riservata a tumori piccoli (inferiori a 4 mm) e si effettua con laser a diodi dopo somministrazione indovena di carboplatino. la termoterapia transpulìpillare ha lo scopo di aumentare la temperatura all’interno della neoplasia e agire in sinergismo con carboplatino, permettendone una migliore captazione da parte delle cellule neoplastiche. Il follow-up di un tumore in trattamento non richiede schemi rigidi per la valutazione dei quadri di regressione. La valutazione della regressione si basa su rilievi oftalmoscopici non definitivamente codificati e rappresenta uno degli aspetti più complessi del trattamento del retinoblastoma, dove è di fondamentale importanza la lunga esperienza dell’oncologo oculare. La radioterapia con sorgente esterna (o teleterapia) è utilizzata nel trattamento del retinoblastoma per la nota radiosensibilità di questo tumore. In realtà, la percentuale di guarigione con la sola teleterapia oscilla, a seconda delle casistiche, tra il 36 e il 69 % dei casi trattati. La discrepanza osservata tra radiosensibilità e percentuale di guarigione stabile è spiegabile, in primo luogo, sulla base dei problemi tecnici che il trattamento comporta, richiedendo l’irradiazione di tutta la retina a dose omogenea e, contemporaneamente, la protezione, per quanto possibile, delle strutture radiosensibili del segmento anteriore dell’occhio (congiuntiva,cristallino e cornea). Tale obbiettivo, in realtà molto difficile da raggiungere, ha ridotto lo sviluppo di tecniche sempre più sofisticate, che si differenziano a seconda dell’incidenza del campo di irradiazione o dell’energia utilizzata in: tecniche a campo anteriore con lente a contatto di protezione del cristallino o a campi laterali con protezione anteriore. La radioterapia esterna è indicata nelle seguenti condizioni: tumori di grandi dimensioni, adiacenti al nervo ottico o papillo-maculari, che non hanno risposto a trattamenti precedenti di chemioriduzione combinata con termoterapia; diffuso seeding vitreale; recidive che non rispondono a nessun altro trattamento conservativo. La radioterapia esterna sull’orbita anoftalmica è indicata in casi di infiltrazione del nervo ottico fino al margine di sezione chirurgica dello stesso, di infiltrazione profonda della sclera e di recidive orbitarie, associata naturalmente a chemioterapia. I quadri di repressione, in seguito a radioterapia esterna, sono i seguenti: Tipo 0: completa scomparsa del tumore (di solito si osserva per tumori molto piccoli, inferiori a 2 mm di diametro) Tipo 1: formazione di una massa completamente calcificata che ricorda il "cottage cheese" (Fig. 7). Tipo 2: formazione di una massa opalescente, traslucida. tipo "fish-flesh; questo è estremamente difficile da differenziare dal tumore ancora attivo. Tipo 3: rappresenta la combinazione dei due precedenti quadri di regressione (1+2). Tipo 4: formazione di una cicatrice corioretinica; rara, si osserva di solito dopo fotocoagulazione, crioterapia o placche episclerali. Le complicanze immediate della radioterapia sono: madarosi, congiuntivite e cheratite attinica, iposecrezione lacrimale; più importanti sono le complicanze tardive che comprendono: corioretinopatia ed otticopatia da radiazioni e cataratta radio-ridotta.
RETINOBLASTOMA E ALTRE NEOPLASIE La presenza di un retinoblastoma aumenta significativamente il rischi di sviluppo di altre forme tumorali. Infatti, i pazienti affetti da forme bilaterali e una minima parte di quelli con forme unilaterali, con mutazione germinale, hanno significative probabilità di comparsa di un secondo tumore non oculare che può essere la causa di morte, indipendentemente da un pregresso trattamento radiante. Si tratta più comunemente di sarcomi osteogenici che possono insorgere sia nell’ambito che fuori dell’orbita irradiata (il più frequente è il sarcoma del femore), ma anche di altre varietà di neoplasie maligne (melanoma, feocromocitroma, seminoma, rabdomiosarcoma, tumori epiteliali maligni). Il termine "retinoblastoma trilaterale" è stato coniato nel 1980 per descrivere la presenza, di solito nella zona della ghiandola pineale, di tumori neuroblastici della linea mediana in individui portatori di alterazioni del gene RB1. In conclusione, il retinoblastoma, il retinoma e i secondi tumori extraoculari, rappresentano manifestazioni della diversa espressione del gene RB1. Tratto da ONCOLOGIA OCULARE, "Retinoblastoma e Rabdomiosarcoma", Teodora Hadjistilianou, Dipartimento di Scienze Oftalmologiche dell'Universita' di Siena. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||