RADICAL
THERAPY OF THALASSAEMIAS
GLAUCO
TORLONTANO (a,b,c),PAOLO DI BARTOLOMEO(b),GABRIELE DI GIROLAMO (b), PAOLA
OLIOSO (b),GABRIELE PAPALINETTI (b), PASQUA BAVARO (b),DONATO NATALE(b),
CESARE BOSMAN (c,d)
(a)
Università «Gabriele D'Annunzio» di Chieti‑Pescara (Italy);(b)
Dipartimento di Ematologia, Terapia Intensiva ‑ Centro Trapianti Midollo
Osseo ‑ Ospedale Civile ‑ Pescara (Italy);(c) Ospedale «Casa
Sollievo della Sofferenza» (IRCCS) ‑ San Giovanni Rotondo (Foggia)(Italy);(d)
Cattedra di Anatomia Patologica ‑ Università «La Sapienza» ‑
Roma (Italy).
*
Estratto da Atti Convegno Lincei “The Thalassemic
Syndromes: A Symposium in honour of Ezio Silvestroni and Ida Bianco.Roma
13.dicembre 1999.
At present, bone marrow transplantation (BMT) can be considered the
treatment of choice for young homozygous thalassaemic patients who have as
donor an unaffected HLA‑identical sibling. However, the possibility of
cure regards only 30% or less of patients in myeloablative BMT with HLA‑identical
sibling donor. The transplant procedure must be considered ethically
acceptable only in presence of low risk of death. In fact, the conservative
trans‑fusion and iron chelating therapy has improved progressively the
quality of life and survival. In Seattle, Thomaset al. considered possible and
acceptable in thalassaemia a 10% mortality risk with 90% post transplant
survival and 75% recovery, on selected very young patients, scarcely or not
yet transfused without evident signs of damaged organs. The first transplant
was successfully realized in 1981 by Thomas, in «a good risk» patient,
managed with few transfusions, an Italian thalassaemic 16 month old boy from a
6yr old HLA‑identical normal sister [1].
The Seattle protocol for graft included:
1. High dose intravenous Methyl‑Busulphan (BU) and moderate doses of Cyclophosphamide (CY), for host immunosuppression and eradication of uderlying disease and creation of «marrow space»;
2. Stem cell graft for rescue from regimen related myelosuppression and
establishment of normal hemopoiesis;
3. Post transplant Methotrexate (MTX) for induction of host‑versus‑graft
tolerance and prevention of severe graft‑versus‑host disease (GvHD).
The
Seattle initial experiment was interrupted after 4 other transplants where 2
children died. After
Seattle, Lucarelli in Pesaro transplanted 13 patients with homozygous
thalassaemia from identical sibling donors. According to Lucarelli a variety
of preparative regimens was utlized involving high doses BU and/or CY and/or
Total Body Irradiation (TBI). MTX was
given after transplant. There were difficulties of eradication of
Thalassaemia,relevant toxicity of treatment and high mortality (46%) [2].
Subsequently, Lucarelli encountered difficulties even with high doses of BU
(16mg/K2) and without TBI. Only afterwards following the Pescara‑London
criteria [3, 5,7] utilizing
Cyclosporin (CyA) and moderate doses of BU he obtained better results [8].
From 1976, year in which our BMT activity started in Pescara, our major
aim has always been to reduce the transplant related mortality and enhance the
clinical and supportive management of the transplanted patients[3].
The progressively decreasing risk related to the procedure encouraged us to
evaluate BMT also in haematological diseases such as thalassaemias,
characterized by several decades of acceptable quality of life with
conservative therapy.
Our BMT program in
Thalassaemia Major was planned in 1981 with a selection of «good risk»
children. Finally in 1983, Barrett in London [4] in collaboration
with our team in Pescara [5,7], began to
transplant some homozygous thalassaemic «good risk» young patients. We were
the pioneers in utilizing moderate doses of BU‑ CY‑ Cy and
Acyclovir (Acv) protocol (Table 1):
•
CY with moderate doses of BU to reduce hepatic toxicity;
•
CyA because we considered it more effective than MTX in post transplant
treatment
for induction of host‑versus‑graft tolerance and
prevention of severe GvHD;
•
Acyclovir to reduce the risk of severe Herpes viruses including CMV infection.
This
regimen demonstrated to be the most effective.
After some successful transplants, Barrett transferred to Bethesda
(USA) and the London (Westminster Hospital) experiment with Thalassaemic patients
was interrupted. After the Pescara/London experience (1983), Lucarelli in
Pesaro in 1985, utilized in Thalassaemia an analogous protocol [8].
In Pescara, patient selection for transplantation has always been very
important. In the initial period (1983‑1987), patients were rigorously
selected, according to: good general and
organic
conditions, without severe iron overload <10 mg/g liver dry); hemoglobin
level generally maintained with transfusions at 9‑10 gr/dl; absence of
severe chronic hepatitis; left ventricular ejection fraction >55% (6‑7,9).
TABLE
1 ‑ Marrow transplantation protocol for Thalassaemia.

During this period we
transplanted 30 patients (10-55 months of follow‑up) with the following
results: Survival 96%; DSF 93%;
Alive with Thalassaemia 4%; Deceased 4% (7,9).
From 1983 till 1986 in Pescara we transplanted only patients up to 8
yrs, in 1987 up to 13 yrs old and since 1988
adult patients as well. These results encouraged us to transplant
progressively older patients. Therefore on February 2, 1988, we transplanted the first adult patient, a 21 yr old woman.
This was probably the first adult Thalassaemic patient in the world cured and
at present, alive and well after a 12 yr follow up period. After this
successful experience our BMT activity was extended also to adult patients and
our policy was to exclude cirrhotic and cardiopathic patients with left
ventricular ejection fraction <55%. However we cautiously admitted to the
protocol patients with relevant iron overload, siderosis, fibrosis and chronic
hepatitis. In these cases we offered the option of BMT only after pre‑transplant
intensification of i.v. chelation therapy and/or following treatment with
Interferon to enhance the chronic hepatitis, if present.
The success of our transplants, other than the choice of the less toxic
myeloablative regimen possible, was determined by the pre‑peri and post
transplant careful management of patients with its progressive improvement.
From May 1983 to June 1.998, we
have performed from HLA identical sibling donors 115 transplants of which 4
second transplants in 111 Thalassaemic patients and with 3 patients off
protocol (1 patient with insufficient doses of BU still living with
Thalassaemia and 2 patients with cirrhotic evolution of the liver) (table
2) [10)
Graft rejection occurred in 4 patients who underwent a second transplant.
Two of these transplanted from the same donor, after immunosuppressive
conditioning with CY and ALG are now alive and disease free 9 and 11 years
respectively after the 2nd BMT.
In conclusion, as of December 1999 on the total of 108 Thalassaemic
transplanted patients, the actuarial probabilities of survival and disease
free survival from a minimal of post transplant follow up of 18 mths to 16 yrs,
are 91% and 87% respectively with 4% alive with Thalassaemia and 9% mortality
(Fig. 1).
Of particular interest in
our case group is also the reduced number of
rejection. This favourable result can be attributed to:
I. hypertransfusional treatment during the two weeks pre‑transplant
period, in order to reduce marrow mass and thus favor myeloablation;
2. favorable take likely due to the always elevated number of marrow
cells constantly infused.
Moreover, the extreme
rarity of rejection in our adult patients should be attributed to the noted
reductions of the marrow mass in the adult thalassaemic with consequently
easier myeloablation.
TABLE
2 ‑ Thalassaemic patients
transplanted in Pescara.

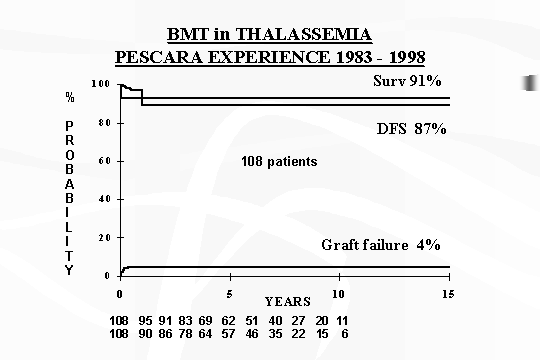
Fig.
1 ‑ Thalassaemia major: survival and disease‑free survival.
The following points of clinical management of BMT in thalassaemia are
for us essential and are still applicable.
1)Hypertransfusion during the 2 weeks prior BMT to reduce the marrow
mass and enhance myeloablation;
2)Accurate skin and mucosal decontamination to reduce the risk of endogenous
infection and severe GvHD and rigorous isolation in «laminar air flow» (LAF);
3)High number of infused nucleated cells: possibly >5x108
in order to favour a stable take. In order to assure adequate doses of marrow,
many patients were also transplanted with donor cryopreserved marrow, in
addition to fresh cells;
4)Careful microbiological monitoring of the patients with prophylaxis
possibly with specific anti‑bacterial, anti‑mycotic and anti‑viral
drugs;
5)Enhancement of clinical and supportive treatment with progressive
risk reduction for severe GvHD and fungal and CMV infections;
6)Moderate dose BU (13‑14 mg/kg) to reduce the overall toxic
effect of the liver; CyA, at least for one year, alone or initially associated
with short course MTX to prevent rejection and severe acute and/or chronic
GvHD;
7) Very prolonged treatment with Acyclovir (9 months post‑BMT) to
reduce the risk of severe Herpes virus infections;
8)Careful management in the first 3 months post
BMT and general protection
during the first year post‑transplant;
9)Exclusion of chelation, which increases the growth of siderofilic microbes,
possibly during the first year post BMT; and, if necessary, preferably use of
early systematic phlebotomy for gradual reduction of iron overload. In effect
the residual post‑transplant immunosupression, relevant especially during
the first year, increases the risk of severe and life‑threatening
infections, especially of siderofilic microbes as Yersinia, Pnemocystics
Carinii and Picornomycosis:
10) Psychological assistance.
In 1990 Lucarelli et al. published the results of 222 young patients (age
<l6 yrs) transplanted between 1983 up to 1989 from HLA identical sibling
donors. After one year post transplantation, the survival and DFS were 82% and
75% respectively with 8% rejection and 18% mortality [8).Only
in 1985, Lucarelli initiated a transplant regimen similar to the Pescara
protocol with moderate doses of BU and CyA starting from day ‑1 to 365
days post transplant [8]. In the first 99 young patients
transplanted from 1985, <16 yrs, the outcome of transplantation was
examined retrospectively in an attempt to identify eventual prognostic
parameters. Three factors seemed to be associated with significantly reduced
probability of survival and disease free survival: poor quality of chelation;
presence of hepatomegaly more or less 2 cm; presence of portal fibrosis.
Therefore three classes were individualized: Class 1-patients with no
factors, characterized by survival and disease free survival 95% and 90%
respectively; Class 2‑patients with only one risk factor and survival
and disease free survival 85% and 81% respectively; Class 3‑patients
with hepatomegaly, liver fibrosis and survival and disease free survival 78%
and 54% respectively.
Subsequently, these results were substantially confirmed from wider cases
reported, but showed less satisfactory results with this protocol and also its
variations, in all adult patients and also in young patients belonging to the
third class [11]. In effect, in 109 adult thalassaemic
patients aged more than 16 yrs, transplanted in Pesaro from HLA‑identical
sibling donor, from 1988 to 1997, the results were the following: Survival
67%, DFS 63%, Mortality 35%, with different protocols for 2nd and 3rd
class patients.
The above classification merits some considerations: 1) the degree of
splenomegaly appears scarcely measurable on physical exam; 2) difficulty in
quantifying the degrees of chelation and the compliance, based on the patient's
history; 3) easier to quantify are the fibrosis and siderosis studied by
histochemical methods. In summary, only the iron overload siderosis and the
fibrosis appear adapt to a quantitative type of evaluation.
As published in the Annals New York Academy of Sciences (Lucarelli, [11]),
Pescara Center came second only to Pesaro for the number of transplants but
not for the quality of the results.
As mentioned before, today in Pescara on the total of 108 transplanted
thalassaemic patients, aged 11 months to 26 yrs, the actuarial probability of
survival and disease free survival, after 16 yrs of experience are the following:
Survival 91%; DFS 87% ; rejection 4%; mortality 9% (Fig. 1).
In the 30 adult patients aged 16‑26 yrs, the results are similar to
those obtained in the global casistics with: Survival 90%; DFS 87%; rejection
3% (1 patient donor was HLA identical uncle); mortality 10%.
Therefore the age of the transplanted
patients (11 months to 26 yrs) did not significantly influence the transplant's
outcome. However, if we consider the age with a take off at 10 yrs old, the
trend seems more favorable for patients under 10 yrs (Fig.
2). This is easily understood since younger patients, in respect to older
patients are less exposed to iron overload, anemia and infections, especially
hepatitis. These damages are greater
in Thalassaemics who
Fig.
2 ‑ BMT inThalassaemia ‑ Pescara experience 1983 ‑ 1998.
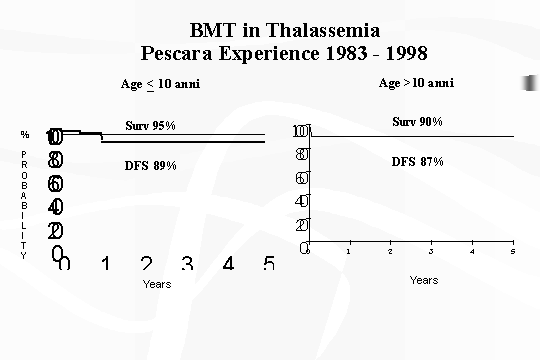
received
less effective conservative therapies. In effect, beyond the frequent virus
hepatitis from multiple transfusions, the most common damage in Thalassaemic
patients is induced by iron loading responsible for lipid peroxidation of
lysosome membranes, followed by release of acid hydrolase into cytoplasm and
finally by cell death [12]. The long term clinical
consequences of this process are: heart failure, liver fibrosis, cirrhosis and
endocrinopathies [13, 14]. Principal
causes of death in our patients were: heart and liver failure, encephalopaty,
GvHD, pneumonia and VOD. According to our experience, fibrosis and chronic
hepatitis singularly do not seem so decisive as cause of mortality from
transplant.
The transplant risk in our patients seemed particularly relevant in adolescent
and adult subjects with serious iron overload due to siderosis and fibrosis,
associated to severe chronic active hepatitis. In fact, with the passing of
the years, this situation increases the damage of iron overload and that of
chronic infections. lf we consider the patients who underwent a liver biopsy
with histochemical studies: of 11 patients (10% of total cases) characterized
by the above mentioned most unfavorable conditions, 4 of them deceased.
While
in the remainin73 patients, only
7 deceased. This difference of mortality is statistically significant (P=
0.03) (Fig. 3).
Our study documents the feasibility of curing Thalassaemia Major with
BMT from an HLA identical sibling, maintaining, also in adult patients, the
transplant related mortality under 10% with disease‑free near 90%.
It is of particular noteworthy that our patients, independently of age,
received a1ways the same protocol for transplantation.
The continuous enhancement in clinical and supportive treatment has allowed
the progressive reduction of risks of severe GvHD, interstitial pneumonia
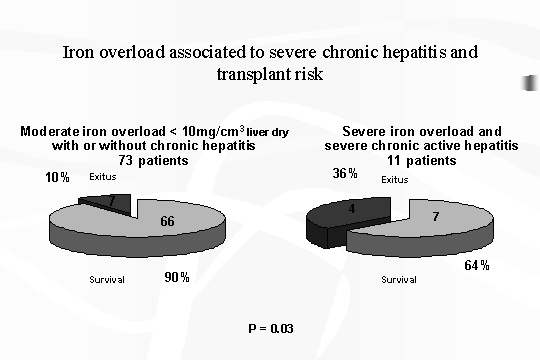
Fig.
3 ‑ Iron overload associated to severe chronic hepatitis and
transplant risk.
and
fungal and cytomegalovirus infections. In fact, in the last 6 years, from June
1992 to June 1998, our results have been particularly favorable [15].
Of the 47 transplanted patients (of which 22 adults) in our protocol, the
survival and disease‑free survival have been 96% and 94% respectively,
with rejection in only 1 patient (transplanted from HLA‑identical uncle)
and with 2 patients deceased (Fig. 4). Of particular
interest is also the reduced number of rejections [15].
The problem of post‑transplant persistence of iron overload is
very important. Our experience and that of other authors document the efficacy
of phlebotomy in iron overload patients following sustained stable engraftment.
Above all, in well iron depleted patients, after this treatment, it is also
possible to increase the eventual response to interpheron therapy given to
improve the chronic hepatitis. Instead early post‑transplant chelation
can increase, according to our experience, the risk of life‑threatening
infections by syderofil microbes, such as Yersinia, Pneumocystis Carinii,
Picomomycetes [16].
NEW DIRECTIONS
1.
Marrow Transplantation with matched unrelated donor or mismatched related
donor
Myeloablative BMT by compatible donor is a curative therapy only for
30% or less of Thalassaemic patients. Recently, relevant progress has been
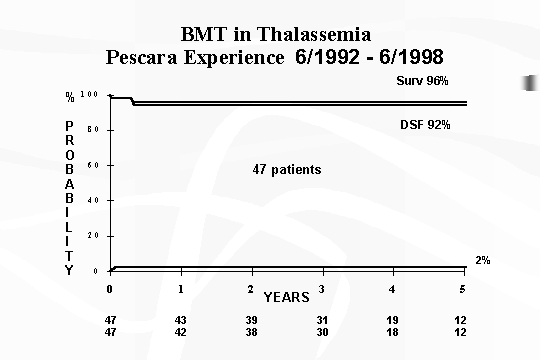
Fig.
4 ‑ BMT in Thalassaemia ‑ Pescara experience 6/1992 –
6/1998.
made
in alternative donor transplantation, thanks to improved methods of HLA typing
for donor selection and for the control of GvHD and the prevention and cure
of fungal and Cytomegalovirus infections [17]. This should
be particularly important to realize BMT in Thalassaemics even in absence of
an HLA‑identical related donor early in the onset of non compliance or
otherwise in case of interruption of chelation therapy. However, further
prudent studies are necessary and should be only within the context of a well
defined research environment such as GITMO
or similar groups. These studies could be realized with less difficulty
within a genetically homogeneous population such as found in Sardinia,
according to Cao [ 11 ] and Contu [ 11
2.
Mini‑transplant non‑myeloablative
low risk regimen
In order to reduce the risk due to toxicity from the actual transplant
program, Storb [ 18] and Slavin [ 19]
have considered the possibility of obtaining the goal of a satisfying
treatment with less toxic non myeloablative protocols in order to realize a
mixed donor‑host chimerism with clinical control of Thalassaemic
disease.
Conditioning regimen of non myeloablative transplantation for its
scarce toxicity would have the advantage of being administered to patients who,
for reason of age limit, or of phases of disease, could not tolerate a BMT
with myeloablative conditioning. Preliminary data of Slavin [19]
seem to suggest that non myeloablative conditioning including Fludarabine,
anti‑T‑lymphocyte globulin and low doses of Busulphan is extremely
well tolerated. At present, besides several patients with malignant
pathologies, also 6 patients with congenital non malignant disorders,
including 1 Thalassaemic, treated with non myeloablative regimen and with
mixed chimerism seem to be alive and well.
Recently, Nagler et al. [20], on the basis of their clinical
experiences, believe that non‑myeloablative conditioning precells of
peripheral blood stem cell transplantation (PBSCT) in connection with donor
lymphocyte infusion (DLI) post allo‑PBSCT, is a promising approach
towards increasing the success rate and optimizing BMT especially in
children with genetic diseases even at an advanced stage of disease when
conventional myeloablative BMT is too risky.
Therefore, pre‑clinical experiments
and clinical observations have led to the development of radically new
concepts in which the myeloablative toxic pre‑transplant conditioning
regimen may be replaced with minimally toxic non‑myeloablative regimens.
Intense temporary immunosuppression before transplantation may be accomplished
also by biological agents, e.g. monoclonal antibodies to surface determinants
on T‑cells, complemented by postgrafting immunosuppression directed
preventing HvG and GvH reactions.
3.
Cord blood stem cell transplantation
After the first experience of Gluckman et
al. [21], the possibility and the utility of
transplantation with Cord Blood Cells (CBCT) are well known today and
evident that this method has less tendency to develop severe GvHD. But at
present, the actual methods of collection have allowed to obtain only in
10‑20% of cases, according to our experience, a sufficient number of
stem cells for the «take» in subjects who weigh over 50 Kg. Therefore, it
seems necessary to optimize the methods of cord blood cells collection and /
or to utilize «ex‑vivo» expansion of these stem cells.
The
GITMO‑GRACE data have shown success in non neoplastic diseases superior to
those in neoplastic ones; moreover, American data document good results also
in transplants from non siblings HLA‑mismatched up to 3 loci [22].
At
present, the Eurocord data regarding CBCT in beta Thalassaemia are not
enthusiastic. In effect, of 12 beta Thalassaemic patients transplanted, all
are alive with the exception of 5 who rejected [23].
Therefore it is necessary to prudently collect with more extensive experience
and overall increase the quantity of injected stem cells.
4.
In utero transplantation in absence of
matched sibling donor
In absence of a matched sibling donor, the «in utero» transplantation
could be an alternative to post‑natal transplantation for the treatment
of congenital haematological disorders, such as Thalassaemia, that can be
diagnosed early in gestation. But until now, according to Make and Zanjani [24],
the treatment of haemoglobinopathies and thus also Thalassaemia, has been unsuccessful
because this approach has not been adequately tested.
According to Flake and Zanjani [24], a winning
strategy could include: early in utero transplant also with minimal chimerism
from parent donor with consequently donor specific tolerance induction; post
natal non myeloablative transplantation from same haploidentical donor.
Of interest is our case of a Thalassaemic child who underwent an «in
utero» transplantation in Palermo with blood from her normal biovular twin
sister. However, this attempt failed and the child was born with Thalassaemia.
Subsequently, at two years of age, at the request of the Palermo Center, the